Cauze Sindromul Alport si simptome, diagnostic si tratament al sindromului Alport
Sindromul Alport (glomerulonefrita familie)conținut
Boala a fost descrisă pentru prima dată de către medic britanic Arthur Alport în 1927.
Sindromul Alport este foarte rar, dar în SUA, este responsabil de 3% din cazuri de insuficiență renală în stadiu terminal la copii și 0,2% dintre adulți, și este considerat cel mai frecvent tip de jad de familie.
Moștenire Sindromul Alport pot fi diferite:
• X-legat dominant (XLAS): 85%.
• autozomal recesiva (ARAS): 15%.
• autozomal dominant (ADAS): 1%.
Cea mai comună formă X-legate de sindromul Alport conduce la stadiu terminal insuficienta renala la barbati. Hematuria apare de obicei la baieti cu sindrom Alport în primii ani de viață. Proteinuria este de obicei absentă în copilărie, dar această condiție de multe ori se dezvolta la barbatii cu XLAS si la ambele sexe cu ARAS. Pierderea auzului si boala de ochi nu a detectat la nastere - apar in copilarie sau adolescenta tarziu, cu mult înainte de dezvoltarea insuficienței renale.
Cauzele și mecanismul de dezvoltare a sindromului Alport
Sindromul Alport este cauzata de mutatii ale genei COL4A4, COL4A3, COL4A5, sunt responsabile pentru biosinteza colagenului. Mutațiile în aceste gene perturba normale colagen de tip IV, care este o componentă structurală foarte importantă a membranelor bazale in rinichi, ochi si urechea interna.Basal membrană - acest subțire structuri de film care susțin tesut separate unul de altul. Când violarea sinteza colagenului de tip IV din membrana bazală glomerulară din rinichi nu sunt în mod normal pot filtra produse toxice din sânge care curge in proteina urina (proteinurie) și eritrocite (hematurie). Anomalii ale sintezei de colagen de tip IV conduce la insuficiență renală și insuficiența renală, care este principala cauză de deces în sindromul Alport.
clinică
Hematuria - este manifestarea cea mai frecventă și precoce a sindromului Alport. hematurie microscopica este observată la 95% dintre femei și aproape toți oamenii. Băieți hematurie este de obicei detectat în primii ani de viață. Dacă băiatul pentru primii 10 ani de viață nu este găsit hematurie, experții americani recomanda să ia în considerare că este puțin probabil ca prezența sindromului Alport.Proteinuria în copilărie este de obicei absentă, dar uneori se dezvolta la baieti cu sindrom Alport X-legate. Proteinurie progreseaza de obicei. proteinurie semnificativă la pacienții de sex feminin este mai puțin frecvente.
Hipertensiunea este adesea prezentă la pacienții de sex masculin cu XLAS și la pacienții de ambele sexe, cu ARAS. Frecvența și severitatea hipertensiunii arteriale crește odată cu vârsta și progresia insuficienței renale.
Pierderea de auz neurosenzorială (pierderea auzului) - este o manifestare caracteristică a sindromului Alport, care apare destul de des, dar nu întotdeauna. Există familii întregi cu sindrom Alport, care suferă de boli renale severe, dar au auz normal. afectarea auzului nu este detectată la naștere. Bilateral de înaltă frecvență pierdere de auz neurosenzorială se manifestă de obicei în primii ani de viață, sau în adolescenta timpurie. Într-un stadiu incipient al bolii este afectarea auzului determinată numai la audiometrie.
Cu progresia, tulburări de auz se extinde la cele mai mici frecvențe, inclusiv vorbirea umană. După apariția pierderii auzului ar trebui să fie de așteptat de implicare rinichi. oamenii de știință americani susțin că, atunci când X-linked sindrom Alport, 50% dintre bărbați suferă de pierdere de auz neurosenzorială în 25 de ani, și timp de 40 de ani - aproximativ 90%.
lenticonus Front (bombarea porțiunea centrală a lentilei ochiului înainte) observate la 25% dintre pacienții cu XLAS. Lenticonus nu este la naștere, dar de-a lungul anilor aceasta duce la pierderea vederii progresiva, care determina pacientii sa schimbe frecvent puncte. Condiția nu este însoțită de dureri în ochi, roșeață sau tulburări de vedere color.
Retinopatia - este cea mai frecventa manifestare a sindromului Alport din partea organului de vizibilitate, afecteaza 85% dintre barbatii cu forma X-linked a bolii. Apariția retinopatiei precede de obicei, insuficienta renala.
Spate polimorfa cornean distrofie - afectiune rara cu sindrom Alport. Cele mai multe nu au nici o plângere. L1649R mutatie in gena colagen COL4A5 poate provoca, de asemenea, o subțiere a retinei, care este asociat cu sindromul Alport X-linkat.
Leiomyomatosis difuz esofagian si arborele bronsic - o alta afectiune rara care apare in unele familii cu sindrom Alport. Simptomele apar in copilarie tarzie si includ deglutiție (disfagie), vărsături, durere epigastrică și în spatele sternului, bronșită frecventă, dificultăți de respirație, tuse. Leiomyomatosis confirmate prin CT sau RMN.
formă autozomal recesivă a sindromului Alport
Pe ARAS reprezintă doar 10-15% din cazuri. Această formă apare la copii ai căror părinți sunt purtători de una dintre gene afectate, a căror combinație provoacă boala la un copil. Părinții nu au simptome sau au simptome minore, iar copiii sunt foarte bolnavi - simptomele lor se aseamănă cu XLAS.formă autozomal dominantă a sindromului Alport
ADAS - aceasta este o forma rara a sindromului, care afectează o generație după alta, și bărbații și femeile care suferă la fel de greu. Manifestările renale și surditate sunt XLAS care amintesc, dar insuficiența renală poate să apară mai târziu în viață. Manifestările clinice ADAS tendință suplimentată la sângerare, makrotrombotsitopeniey, sindromul Epstein, prezența neutrofilica incluziunilor în sânge.Diagnosticul sindromului Alport
• Teste de laborator. Analiza urinei: pacienții cu sindrom Alport, sânge în urină (hematurie) apare cel mai frecvent, și un conținut ridicat de proteine (proteinurie). Analizele de sânge demonstrează insuficiență renală.• Un tesut biopsie. țesut de rinichi obținut prin biopsie au fost examinate prin microscopie electronică pentru prezența anomaliilor ultrastructurale. O biopsie de piele este mai putin invazive, iar experții americani recomanda o fac în primul rând.
• Analiza genetică. In diagnosticul de sindrom Alport, în cazul în care există îndoieli după o biopsie de rinichi, analiza genetica este folosit pentru a obține un răspuns clar. Definită genei de colagen de tip IV mutație.
• audiometrie. Toți copiii cu istoric familial sugestiv de sindrom Alport, ar trebui să se supună audiometrie de înaltă frecvență pentru a confirma pierderea de auz neurosenzorială. Se recomandă monitorizarea periodică.
• examen oftalmologic. Examinarea de către un oftalmolog este important pentru depistarea precoce și monitorizarea lenticonus față și alte anomalii.
• ecografie renală. În etapele ulterioare ale sindromului Alport, ecografia renala ajuta la identificarea anomalii structurale.
Experții britanici, pe baza de date noi (2011) privind mutațiile genetice la pacienții cu sindrom X-linked Alport recomanda de testare pentru mutatii genetice COL4A5, în cazul în care pacientul întrunește cel puțin două criterii de diagnostic pentru Grigorie, și COL4A3 și analiza COL4A4 dacă COL4A5 mutație nu detectate sau suspectate de moștenire autosomal.
Tratamentul sindromului Alport
Sindromul Alport este incurabilă până în prezent. Studiile au arătat că inhibitorii ECA pot reduce proteinuria și încetinește progresia insuficienței renale. Astfel, utilizarea inhibitorilor ECA la pacienții cu proteinurie expedient, indiferent de prezența hipertensiunii arteriale. Același lucru este valabil și pentru antagoniști ai receptorilor de ATII. Ambele clase de medicamente pare a ajuta la reducerea proteinurie prin reducerea presiunii intraglomerular. Mai mult, inhibarea angiotensinei II, factor de creștere responsabil pentru scleroza glomerulară, poate retardatule teoretic de întărire.Unii cercetători sugerează că tacrolimus este capabil de a reduce proteinurie și stabilizarea funcției renale la pacienții cu sindrom Alport (studii au fost mici). Dar rapoarte sugereaza ca raspunsul pacientilor la ciclosporină este foarte variabilă, și, uneori, medicamente poate precipita fibroza interstițială.
În insuficiența renală, terapiile standard includ eritropoietina pentru a trata anemia cronică de medicamente pentru a controla osteodistrofia, corectarea acidozei si medicamente antihipertensive, pentru controlul tensiunii arteriale. hemodializă aplicată și dializă peritoneală. Pacienții cu sindrom Alport, transplantul de rinichi nu este contraindicat: Experiența de transplant în Statele Unite, a demonstrat rezultate bune.
Terapia genică pentru diferite forme de sindrom Alport este o optiune promitatoare de tratament, care este acum în mod activ de a fi studiate laboratoare medicale occidentale.
Distribuiți pe rețelele sociale:
înrudit
 Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus
Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus Akromezomelicheskaya displazie. sindromul eykardi
Akromezomelicheskaya displazie. sindromul eykardi Asistență medicală de urgență priprogressiruyuschem rapid glomerulonefrita
Asistență medicală de urgență priprogressiruyuschem rapid glomerulonefrita Patogeneza insuficienței renale cronice. Cercul vicios al insuficienței renale
Patogeneza insuficienței renale cronice. Cercul vicios al insuficienței renale Boala autozomal dominanta de rinichi polichistic la copii. Diagnostic si tratament
Boala autozomal dominanta de rinichi polichistic la copii. Diagnostic si tratament Boala autozomal recesiva de rinichi polichistic la copii. Diagnostic si tratament
Boala autozomal recesiva de rinichi polichistic la copii. Diagnostic si tratament Rapid progresivă (fasceita extracapillary) glomerulonefrita. Diagnostic si tratament
Rapid progresivă (fasceita extracapillary) glomerulonefrita. Diagnostic si tratament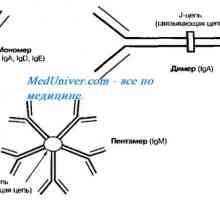 Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation
Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation Insuficiență renală acută la copii. motive
Insuficiență renală acută la copii. motive Sindrom nefrotic secundar și congenitale la copii. diagnosticare
Sindrom nefrotic secundar și congenitale la copii. diagnosticare Insuficiență renală cronică la copii. motive
Insuficiență renală cronică la copii. motive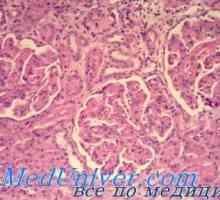 Nefrita tubulointerstițială cronică la copii. Diagnostic si tratament
Nefrita tubulointerstițială cronică la copii. Diagnostic si tratament- Cauzele glomerulonefrită și diagnostic, tratament și complicații ale glomerulonefrita
- Nefrita ereditara. Etiologia și patogenia nu sunt înțelese. Se presupune că boala este asociata cu…
- Sindromul capillaritis sistemic Goodpasture care afecteaza in principal pulmonare si de tip…
- Sănătate Enciclopedia, boli, medicamente, medic, farmacie, infecție, rezumate, sex, ginecologie,…
- Terapie
 Boala cronică de rinichi: stadiu, clasificare, cauze, simptome, diagnostic, tratament, simptome
Boala cronică de rinichi: stadiu, clasificare, cauze, simptome, diagnostic, tratament, simptome Nefropatie ereditară
Nefropatie ereditară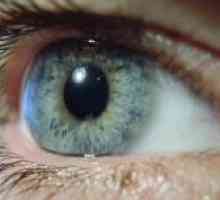 Defecte congenitale de noapte și vederii periferice
Defecte congenitale de noapte și vederii periferice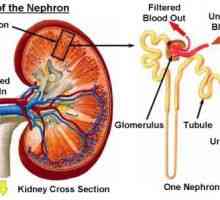 Glomerulonefrită, tratament, simptomele, cauzele
Glomerulonefrită, tratament, simptomele, cauzele
 Akromezomelicheskaya displazie. sindromul eykardi
Akromezomelicheskaya displazie. sindromul eykardi Sindromul Bartter Sindromul și Gitelman la copii: simptome, tratament, cauze
Sindromul Bartter Sindromul și Gitelman la copii: simptome, tratament, cauze Nefrita tubulointerstițială cronică la copii. Diagnostic si tratament
Nefrita tubulointerstițială cronică la copii. Diagnostic si tratament Boala autozomal dominanta de rinichi polichistic la copii. Diagnostic si tratament
Boala autozomal dominanta de rinichi polichistic la copii. Diagnostic si tratament Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus
Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus Rapid progresivă (fasceita extracapillary) glomerulonefrita. Diagnostic si tratament
Rapid progresivă (fasceita extracapillary) glomerulonefrita. Diagnostic si tratament Defecte congenitale de noapte și vederii periferice
Defecte congenitale de noapte și vederii periferice Glomerulonefrită, tratament, simptomele, cauzele
Glomerulonefrită, tratament, simptomele, cauzele Sindrom nefrotic secundar și congenitale la copii. diagnosticare
Sindrom nefrotic secundar și congenitale la copii. diagnosticare Boala autozomal recesiva de rinichi polichistic la copii. Diagnostic si tratament
Boala autozomal recesiva de rinichi polichistic la copii. Diagnostic si tratament