Boli lizozomale de la copii

conținut
Multe dintre mai mult de 40 descrise boli ereditare cu defecte ale enzimelor lizozomale caracterizate de boli hepatice.
Acest lucru se aplică în primul rând pentru boala Gaucher, Niemann - Pick, Wolman, un număr de mucopolizaharidoza, oligosaharidozov, leucodistrofia metacromatică, boala lui Farber, Fabry, gangliosidosis tip I.
Boala Gaucher. Există hepatomegalie. Histologic determinate celule Gaucher, care sunt marite celule Kupffer cu nucleu situat excentric și citoplasmei caracteristic având forma „hârtie absorbantă mototolită.“ In aranjamentul grupelor de celule Gaucher în lobulului și portal tracturile hepatice pot fi marcate fibroza. Se poate dezvolta ciroza.
Boala Niemann - Pick. Cu implicarea în procesul de ficat patologic devine mărit, palid. Microscopic: Structura lobuli nu este rupt, fibroza nu este tipic. Celulele Kupffer a crescut, spuma au vacuolate citoplasmă. Modificări similare au fost observate în hepatocite. In celulele, acumularea de ceroid, colesterol și fosfolipide. Microscopia electronică a arătat figura mielină caracteristică compusă din plăci osmiophilic concentrice sau paralele. In activitatea clinica sfingomielinazei întruchipare boala cu aspectul morfologic normal poate fi observată în ficat și leucocite, astfel hepatita neonatală.
Boala Wolman. Ficatul este mărit, vopsit în galben. Steatoza Histologic are loc cu prezența unui număr mare de celule spumoase, care sunt hepatocite, celule Kupffer, care conțin cantități mari de colesterol și lipide. Colesterolul este cel mai bine evidențiat prin examinarea secțiunilor de țesut hepatic lumină polarizată congelat.
Mucopolysaccharidosis. leziuni hepatice cauzate de acumularea în celulele și substanța intercelulară și glicozaminoglicanilor acide gangliozide simultan. Există hepatomegalie. Histologic manifestata vacuolizarea hepatocite si celule Kupffer și, într-o măsură mai mică, epiteliu duetului biliar. Poate dezvolta in cele din urma fibroza hepatica.
Mucolipidosis. Ficatul este mărit, vopsit în galben. Steatoza Histologic are loc cu prezența unui număr mare de celule spumoase, care sunt hepatocite, celule Kupffer, care conțin cantități mari de colesterol și lipide. Colesterolul este cel mai bine evidențiat prin examinarea secțiunilor de țesut hepatic lumină polarizată congelat.
Oligosaharidozy. Ficatul este implicat în procesul în toate formele de boala. Există hepatomegalie cu vacuolizarea citoplasmei hepatocitelor și a celulelor Kupffer. Vacuole de dimensiuni diferite, pot fi colectate într-o destul de mare. Vacuolizare se poate produce în celulele endoteliale și epiteliul canalului biliar.
leucodistrofia metacromatică. Macrofagele din tracturile portal de ficat determinat granule metacromatice, și ele pot fi, de asemenea, detectate în celulele Kupffer și fibroblaste. Vezica biliara poate fi determinat excrescențe papilare mucoasei la prezența celulelor spumoase în zona subepitelial.
Boala lui Farber. Există hepatomegalie. Acumularea de ceramide și gangliozidelor are loc în hepatocite cu vacuolizarea citoplasmei, prin care acestea dobândesc un aspect spumos. În plus, se dezvoltă un proces granulomatoasă reactiv cu rezultatul fibrozei. Lipogranulemy împrăștiate țesutul hepatic, constau din limfocite, macrofage, celule gigant multinucleate.
Boala Fabry. Ca și în alte organe, acumularea globotriaosiltseramida hepatice are loc în principal în celulele endoteliale vasculare.
Gangliosidosis. In formele de boala care implica ficat apare hepatomegalie. Microscopia Lumina este marcat vacuolizarea citoplasmei hepatocitelor și a celulelor Kupffer, ceea ce le conferă un aspect spumos. Microscopia electronică extinsă la lizozomii determinate transparente, care conțin o sită cu ochiuri de material granulat.
boli de stocare lizozomale
enzime lizozomale degradează macromolecule sau celula în sine (de exemplu, atunci când reciclat componente de celule structurale) sau capturate din exterior. defecte Mostenita sau deficientele enzimelor lizozomale (lizozomale sau alte componente) poate duce la o acumulare a metaboliților nedegradate. Prin prezența a numeroase boli specifice de depozitare, deficientele sunt de obicei grupate în funcție de metabolit biochimică acumulat.
Subgrupuri includ:
- Mucopolysaccharidosis,
- sfingolipidoz (lipidoses)
- Mucolipidosis.
Este cel mai important și Mucopolysaccharidosis sfingolipidoz. Tipul 2 de glicogen boala de stocare este o tulburare lizozomale de stocare, dar de cele mai multe glicogenului - nr.
Deoarece celulele reticuloendotelial (de exemplu, splină) lizozomii bogate, tesutul reticuloendotelial deteriorat de unele boli lizozomale de stocare, dar, de obicei, țesut mai bogat substrat sufera vsego.Takim mod mai puternic, creierul, care este bogat în gangliozide, mai ales care suferă de gangliosidosis, întrucât mucopolysaccharidosis afectează multe țesuturi deoarece mucopolizaharide prezente în organism.
Mucopolysaccharidosis (MPS). Mucopolysaccharidosis - lipsa ereditară a enzimelor implicate în distrugerea glicozaminoglicani. Glicozaminoglicani (cunoscut anterior ca mucopolizaharide) sunt convenționale și cu celule polizaharide de suprafață și structuri ale matricei extracelulare. Deficienta enzimei, prevenind distrugerea acumulării glicozaminoglican, cauza fragmentelor glicozaminoglicani în lizozomi și pot provoca modificări extensive osoase, a țesuturilor moi și ale sistemului nervos central. Mostenirea este de obicei autosomal recesiv (cu excepția mucopolysaccharidosis de tip II).
Vârsta, manifestările clinice și severitatea variază în funcție de tipul.
Manifestările comune includ caracteristici faciale grosiere, întârzie dezvoltarea nervoase și de regresie, contracturile articulare, organomegalie, păr aspru, insuficienta respiratorie progresiva, malformații cardiace, modificări scheletice și subluxație a vertebrelor cervicale.
Diagnosticul este sugerat de istoricul medical, datele examenului fizic, anomalii osoase (de exemplu, dysostosis multiple), au fost detectate în timpul studiului a scheletului și totale și fracționate glicozaminoglicani crescute. Diagnosticul este confirmat prin testul enzimatic în cultura fibroblastelor (prenatală) sau leucocite periferice (după naștere). Teste suplimentare se efectuează pentru controlul modificărilor specifice organelor (de exemplu, defecte de supapă ecocardiografie, audiometrie pentru a detecta schimbări în cadrul ședinței).
Tratamentul de mucopolysaccharidosis tip I (boala Hurler) constă în umplerea enzimei -L-iduronidazy, care se oprește în mod eficient progresia și restabilește toate complicațiile non-SNC ale bolii. Transplantul de celule stem hematopoietice (CSH) sa dovedit eficace in primele studii, dar ineficiente in boala SNC.
Sfingolipidoz. Sphingolipids - componente lipidice normale ale membranelor celulare, se acumuleaza in lizozomii si determina modificari extinse in neuroni, oase și alte modificări atunci când defectul enzima previne distrugerea lor. Desi incidenta este scazuta, unele forme de frecvență purtătoare este mare. Boala Gaucher este cea mai comuna fingolipidozom.
Boala Gaucher
Boala Gaucher este sfingolipidozom, în curs de dezvoltare, ca urmare a deficitului de glucocerebrosidază ceea ce duce la acumularea de glucocerebrozidă și compușii înrudiți. Simptomele și semnele variază ottipa.
Tipul I (neneyropatichesky) este cel mai frecvent (90% din totalul pacienților). Activitatea enzimatică reziduală este maximă. Evreii Ashkenazi sunt cele mai risku- 1/12 sunt purtători. Vârsta de debut a bolii variază de la 2 ani până la sfârșitul vieții de adult. Simptomele și semnele includ hepatosplenomegalie, boala osoasa, retard de creștere, pubertate întârziată, echimoze și PINGUECULA. sângerări nazale și echimoze care rezultă trombocitopenia sunt manifestări comune. Radiografia arată capetele oaselor lungi caracteristice (kolboobraznaya Erlenmeyer de deformare) și subțierea corticală.
Tip II (neuropatia acută). Debutul bolii la perioada de copilarie.
Tip III (subacute neuropatică) asupra morbidității, activitatea enzimelor și severitatea clinică situată între tipurile I și II. Boala apare în orice moment în timpul copilăriei. Manifestările clinice depind de subtipul și includ demența și ataxie progresivă (Ilia).
diagnosticare
- Determinarea activității enzimei.
Diagnosticul se bazează pe analiza enzimelor de leucocite. Transportatorii să identifice și să distingă tipuri de analiza de mutații. Desi biopsia nu este celulele necesare Gaucher - lipide macrofage umplute în ficat, splină sau măduvă osoasă, au formă de hârtie subțire mototolită - sunt diagnostice.
tratament
- În tipurile I și III: replenishing enzimă sau glucocerebrozidaza placentar recombinant.
- Uneori miglustat, splenectomie sau transplantul de celule stem.
Pacienții care primesc substituție enzimatică, necesită monitorizarea regulată a valorilor hemoglobinei și trombocitelor, evaluarea periodică a splinei și starea volumului ficatului, CT sau MRI, precum și evaluări periodice ale stării bolii osoase în studiul scheletic, cu energie duala cu raze X ab sorbtsiometrii sau RMN.
Splenectomia poate fi de ajutor la pacientii cu anemie, leucopenie, trombocitopenie sau atunci când mărimea splinei cauzează disconfort.
Boala Niemann Pick
Boala Niemann - Peak - sfingolipidoz cauzată de activitatea insuficientă a sfingomielinazei, ceea ce duce la acumularea de sfingomielina (fosforilcolină ceramide) celule reticuloendotelial.
Pacienții cu tip A sunt <5% нормальной активности сфингомиелиназы. Заболевание характеризуется гепатоспленомегалией, задержкой развития и быстро прогрессирующей нейродегенерацией.
La pacienții cu activitatea sfingomielinazei de tip B este de 5-10% din normal. Tipul B are o manifestări clinice diferite decât cele de tip A pot dezvolta limfadenopatie și hepatosplenomegalie. Pancitopenie destul de comună. Cei mai mulți pacienți cu tip B au putin sau deloc leziuni neurologice și de a supraviețui la maturitate, ele pot fi distinse clinic la pacienții cu boala Gaucher de tip I. În cazurile severe, cum ar fi în infiltrate pulmonare progresive provoca complicații majore.
diagnosticare
- screening-ul prenatal.
- Studia leucocite sfingomielinază.
Ambele tipuri sunt, de obicei nu cunosc istoricul medical și rezultatele cercetării, în primul rând hepatosplenomegalie. Diagnosticul poate fi confirmată prin analiza leucocitelor sfingomielinazei, iar acest lucru se poate face prin amniocenteză sau prenatally chorionic eșantionare coriale.
tratament
Transplantul de celule de măduvă osoasă sau stem investigate ca posibile optiuni de tratament.
boala Tay-Sachs si Sandhoffa
Tay - Sachs si boala Sandhoffa - sfingolipidozy cauzate de o deficienta de hexozaminidază, provoca simptome neurologice severe si moartea timpurie.
Gangliozide sunt sfingolipide complexe. Există două forme de bază, GM1 și GM2, care pot fi implicate în bolile lizozomale nakopleniya- disting două tipuri principale de GM2 gangliosidosis, fiecare dintre acestea putând fi cauzate de o serie de mutații diferite.
Tay - Sachs boala. Moștenirea este autozomal recesiva, cele mai frecvente mutații detectate în evreiesc (Ashkenazi) coborare in Europa de Est 1/27 adult normal, cu toate că alte mutații detectate în unele populații de populație și cajun franco-canadiană.
Principalele etape de dezvoltare a copiilor cu Tay - Sachs încep să rămână după vârsta de 6 luni, în același timp, în curs de dezvoltare o deteriorare progresivă a funcțiilor cognitive și motorii, ceea ce duce la dezvoltarea de convulsii, retard mental, paralizie si deces la copii sub 5 ani. Roșu-vișiniu erupții cutanate makleznaya este un simptom comun.
In absenta tratamentului eficace este axat pe gestionarea adulților de vârstă reproductivă de screening in grupurile de risc ridicat pentru detectarea transportatorilor (prin testarea activității enzimatice și prezența mutațiilor), în asociere cu consiliere genetica.
boala Krabbe
Boala Krabbe este sfingolipidozom, cauzand retard mental, paralizie, progresează până la moarte.
Boala lui Krabbe (lipidosis galactosylceramide, leukodystrophy celula globoid) este cauzata de un deficit de galactocerebrozidă galactozidaza autosomal recesivă.
Aceasta boala afecteaza copiii si se caracterizeaza prin retard mintal, paralizie, paralizie pseudobulbară, progresiva la moarte.
Deoarece transplantul de măduvă osoasă întârzie în mod eficient apariția simptomelor, este uneori efectuat diagnosticul prenatal sau nou-nascuti de screening de rutina (New York).
leucodistrofia metacromatică
sfingolipidozom leucodistrofia metacromă este asociat cu deficit de arylsulfatase, care provoaca paralizia progresiva si dementa care duce la deces la varsta de 10 ani.
Când metacromă leukodystrophy (sulfatide lipidosis) deficit arylsulfatase determină o acumulare de lipide in materia alba metacromă a sistemului nervos central, nervii periferici, pochkah- se acumuleaza in sistemul nervos determină demielinizarea central și periferic. Există numeroși pacienți mutatsii- separate în funcție de vârstă, începutul și rata de progresie a bolii.
Forma infantil se caracterizeaza prin paralizie progresiva, si dementa, apare de obicei înainte de vârsta de 4 ani și conduce la deces în aproximativ 5 ani de la debutul simptomelor. Forma juvenila începe la vârsta între 4 și 16 ani, cu tulburări ale mersului, tulburări intelectuale și simptome de neuropatie periferică. Spre deosebire de forma infantil, reflexele tendinoase profunde sunt, de obicei ocupat. Există, de asemenea, o formă mai moale pentru adulți.
Diagnosticul este sugerat de simptome clinice si detectarea vitezei reduse de conducere nervoasă.
Tratamentul eficient nu există.
Boala Fabry
Boala Fabry este sfingolipidozom cauzata de deficienta galactozidaza.
Boala Fabry (angiokeratoma Corporis diffusum) este deficiență legată-X a galactozidază enzimei lizozomale, care este necesară pentru catabolismul normală trigeksoziltseramida. Glicolipid (globotria-oziltseramid) se acumulează în multe țesuturi (de exemplu, endoteliul vaselor sanguine, vase limfatice, inima, rinichi).
Diagnosticul la barbati, pe baza apariției leziunilor tipice (angiokeratomas) în partea inferioară a corpului și caracteristicile de neuropatie periferică, opacizare a corneei, și episoade febrile recurente. Moartea este rezultatul insuficienței renale sau cardiace și complicații cerebrovasculare. femeile heterozigote sunt de obicei asimptomatice, dar pot avea o formă slăbită a bolii.
Tratamentul - înlocuirea -galakgozidazoy recombinant enzimă (galactozidaza beta), combinate cu măsuri de susținere în febră și durere. Transplantul de rinichi este eficient în tratamentul insuficienței renale.
colesterol boala de stocare ester și boala Woolman
Colesterolul boalå ester și boala Woolman sfingolipidozami sunt cauzate de deficiența de lipază acidului lizozomale, rezultând în hiperlipidemie și hepatomegalie.
Aceste boli - rare autozomale tulburări recesiva care rezultă din acumularea de esteri de colesterol și de trigliceride, în principal în lizozomi de histiocytes, ceea ce duce la apariția de celule spumoase în ficat, ganglionii limfatici si alte tesuturi.
Boala lui Woolman - o formă severă, se manifestă în prima săptămână de viață, inapetență, vărsături, distensie abdominală, și secundar gepatosplenomegalii- copii mor, de obicei, în termen de 6 luni.
acumularea de colesterol esteri ai bolii este mai puțin severă și se poate manifesta în vârstă, chiar și la maturitate, atunci când se poate demonstra gepatomegaliya- poate dezvolta ateroscleroza prematura, de multe ori grele.
Diagnosticul se bazează pe simptome clinice de deficiență a lipazei acidului semne în probele de ficat biopsie fibroblaste de piele de cultura, limfocite sau alte țesuturi. Diagnosticul prenatal se bazează pe absența activității lipazei acide în vilozități corionice cultivate.
Nu există nici un tratament eficient, dar statine reduce nivelurile de LDL plasmatice.
 Boala Gaucher Glyukotserebrozidoz. Boala Glikosfingolipidoz Fabry
Boala Gaucher Glyukotserebrozidoz. Boala Glikosfingolipidoz Fabry Diagnostic sfingomielinoza. corpurile se schimbă la NPD
Diagnostic sfingomielinoza. corpurile se schimbă la NPD Amavroticheskaya imbecilitate congenitală. Mucolipidosis
Amavroticheskaya imbecilitate congenitală. Mucolipidosis Diagnostic sulfatidoza. Sfingomielinoz boala Niemann Pick
Diagnostic sulfatidoza. Sfingomielinoz boala Niemann Pick Moarte fetală intrauterină cu macerare. Simptomele de boala hemolitica a nou-născutului
Moarte fetală intrauterină cu macerare. Simptomele de boala hemolitica a nou-născutului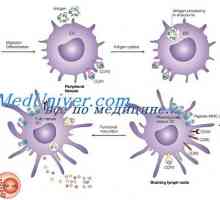 Efectul imunomodulatoare asupra celulelor dendritice. Morfologia celulelor dendritice
Efectul imunomodulatoare asupra celulelor dendritice. Morfologia celulelor dendritice Icter neonatal cu tulburări metabolice
Icter neonatal cu tulburări metabolice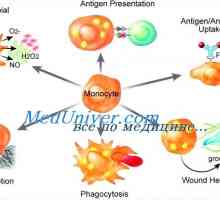 Disfuncția de monocite și macrofage imunitate
Disfuncția de monocite și macrofage imunitate Transplantul de celule stem pentru acumularea de boli si talasemie
Transplantul de celule stem pentru acumularea de boli si talasemie Boli ale sistemului hematopoietic. Boala Gaucher
Boli ale sistemului hematopoietic. Boala Gaucher- Boala Gaucher se referă la boli sfingolipidozam acumulare lipidov- datorită unui defect al genei…
- Sănătate Enciclopedia, boli, medicamente, medic, farmacie, infecție, rezumate, sex, ginecologie,…
- Sănătate Enciclopedia, boli, medicamente, medic, farmacie, infecție, rezumate, sex, ginecologie,…
- Terapia, boala a sistemului digestiv
 Boli lizozomale
Boli lizozomale Violarea metabolismului lipidelor
Violarea metabolismului lipidelor Boala, simptome, tratament Gaucher
Boala, simptome, tratament Gaucher Boala Niemann Pick
Boala Niemann Pick Fibroza hepatică congenitală
Fibroza hepatică congenitală Mucolipidosis
Mucolipidosis Tulburări metabolice ale metabolismului acidului uric și a metalelor
Tulburări metabolice ale metabolismului acidului uric și a metalelor
 Mucolipidosis
Mucolipidosis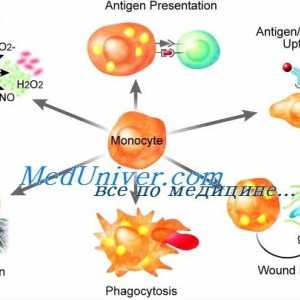 Disfuncția de monocite și macrofage imunitate
Disfuncția de monocite și macrofage imunitate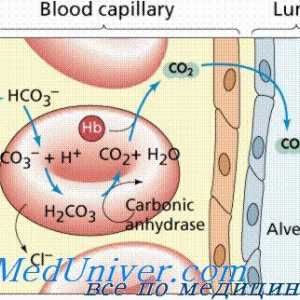 Amavroticheskaya imbecilitate congenitală. Mucolipidosis
Amavroticheskaya imbecilitate congenitală. Mucolipidosis Tulburări metabolice ale metabolismului acidului uric și a metalelor
Tulburări metabolice ale metabolismului acidului uric și a metalelor Boala Niemann Pick
Boala Niemann Pick Violarea metabolismului lipidelor
Violarea metabolismului lipidelor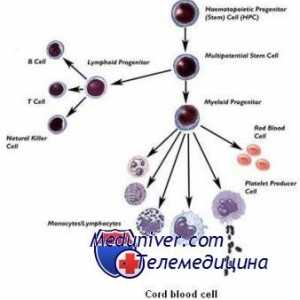 Transplantul de celule stem pentru acumularea de boli si talasemie
Transplantul de celule stem pentru acumularea de boli si talasemie