Consiliere genetică
Cand genetice practica de consiliere standard este de a determina probabilitatea bolii, exprimată ca fracții sau procente. Există mai multe moduri de a privi două estimări de risc: Mendel (genetice) și empirice. Metoda Mendel de evaluare a riscului pentru identificarea tulburărilor care sunt cauzate de modificari ale unei singure gene, cu un tip cunoscut de moștenire. Prin urmare, în condiții care sunt moștenite dominantly, riscul descendența 50%. Dacă un părinte are o tulburare autozomal recesiva, de exemplu, sindromul Ellis van Creveld, riscul este neglijabil. Acest lucru se datorează faptului că, în autosomal boli recesive copilul trebuie să primească gena anormale, și de la tatăl său și de mama. Șansa ca partenerul femeii bolnav este un purtător al unei gene recesive rare este foarte mică, dacă nu este blooded rodstvennikom- ei, în acest caz, riscul pentru copil va fi destul de mare.
Determinarea riscului empiric, mai degrabă se bazează pe observarea și studiul familiilor cu o anumită boală decât pe pozițiile teoretice, ținând cont de tipul de moștenire. se adoptă această metodă pentru cele mai frecvente tulburări non-mendeliană, cum ar fi defecte de tub neural, buza cleft si palatului, precum și pentru majoritatea defectelor cardiace izolate.
conținut
Video: Paradise adevărat și fals 48
Tulburări Mendel
Când consilierea unei femei gravide cu boli cardiace congenitale trebuie să determine mai întâi dacă are un defect cardiac izolat, sau există alte simptome însoțitoare, care se poate presupune tulburare asociată cu o singură genă (mendeliană).
Sindromul Holt-Oram
Combinația de anomalii scheletice ale membrelor superioare cu boli cardiace congenitale (de obicei, defect septal atrial - ASD) a fost descrisă pentru prima dată în 1960 de către Holt și Oram. Aceasta este o conditie autosomal dominanta cu manifestări extrem de variabile din partea scheletului și a inimii.
De obicei suferă degetele, care se manifestă fie hipoplazie sau prezența a trei falangelor sau absența completă a degetelor. Anomaliile poate fi radial, ulnar și oasele humerusului, și, în general, pacientul nu poate efectua supinație și pronație a încheieturii. Uneori există focomelia extremități superioare. membrele inferioare nu sunt afectate.
D M P P - este o anomalie cardiacă care este detectată în două treimi din cazuri. Cu toate acestea, în cazul în care familiile alte rude au avut o semne tipice ale bolii au fost descrise persistenta canalului arterial, coarctație de aortă, VSD, transpunerea vaselor mari, si prolaps de valva mitrala.
Uneori nu există boli cardiace congenitale, dar pot prezenta aritmie sau ECG ușoare modificări.
dizabilități mentale sau dificultăți de învățare nu sunt un semn al sindromului Holt-Oram. ultrasonografie detaliate, inclusiv ecocardiografie fetală trebuie să fie oferite la 18-20 săptămâni de sarcină, pentru a fi în măsură să identifice gradul de anomalii la nivelul membrelor și pentru a discuta despre comportamentul și prognosticul în continuare. defect septal atrial, anomalie a inimii, care este cel mai frecvent în sindromul Holt-Oram, este imposibil să se determine fătul, dar este necesar, pentru a exclude defecte mai rare. gena TVH5 care cauzeaza sindromul Holt-Oram, localizat pe cromozomul 12.
sindromul Noonan
Acesta este un sindrom dismorfny relativ frecvent asociat cu boli cardiace congenitale, stenoza pulmonara de obicei. Există o tulburare autosomal dominanta, dar în aproximativ jumătate din cazuri - sporadice, ele sunt rezultatul unor noi mutații. trasaturile faciale caracteristice sunt adesea mai vizibile în copilărie și la adulți acestea sunt mai greu de recunoscut. Sa constatat că cauza încălcări în jumătate din cazuri este semnalul gena PTPN11 al cromozomului 12, identificarea care poate confirma un diagnostic clinic.
Caracteristici principale Sindromul Noonan - un corp scurt, un gât larg sau membranoasă și deformare toracică (pectus excavatum sau carinatum), asociată cu pleoape caneluri aval, ptoza, urechi set joase dislocate din spate și lombare linia parului. Criptorhidismul este adesea două sensuri, se găsește în 60% dintre băieți și pot necesita tratament chirurgical. La fete, nici o anomalie organele genitale. În 10% dintre copiii cu sindrom Noonan pot avea dificultăți în procesul de învățare. Ca rezultat al anomalii ale căii intrinseci a cascadei de coagulare se poate produce sangerare post-operatorie.
defecte cardiace congenitale apar la majoritatea pacienților. Cele mai frecvente dintre ele - stenoza arterei pulmonare, care pot fi corectate chirurgical. Aproximativ 10% dintre pacienți există o cardiomiopatie hipertrofica, care poate fi însoțită de insuficiență cardiacă în copilarie.
Diagnosticul prenatal se realizează cu ajutorul unor probe CVS, în cazul în care prezența confirmată a mutației în familie. În alte cazuri, atunci când unul dintre părinți are sindrom Noonan, sau în familie, înainte de copii s-au născut cu acest sindrom, prezinta ecocardiografie fetală detaliată.
sindromul LEOPARD
Este o abreviere a numelor engleză caracteristicile esențiale tipice acestui sindrom (L - lentigo, E - modificări ECG O - oculară oftalmic) hypertelorism (P - stenoza pulmonara [stenoza pulmonara] A - anomalii genitale, R -zaderzhka creștere D - surditate neurosenzorială). Acest diagnostic trebuie amintit în prezența pacientului toate aceste semne în combinație cu mai multe lentigo. Sindromul autozomal dominant moștenit, cu diferite grade de exprimare, poate exista o ușoară retard mintal.
În aceeași încălcare a descoperit mutații ale aceleiași gene, și sindromul Noonan, și, aparent, ele se pot suprapune. Consultarea la genetica poate ajuta în planificarea viitoare anchete.
Descrie diferitele schimbări în inimă. ECG-ul poate fi detectată devierea axei, într-o direcție sau două sensuri de hipertrofie sau de conducere tulburari, de exemplu, lungirea intervalului P-R, o blocada parțială, blocarea picioarelor sau blocarea completă. stenoza pulmonară poate fi în valve sau pâlnie. defecte mai puțin obișnuite, cum ar fi stenoza aortica, stenoza mitrală și cardiomiopatie obstructivă.
Dacă un părinte are sindromul LEOPARD, ecocardiografie fetală este prezentată pentru a evita boli cardiace congenitale severe.
Video: Medikogeneticheskoe consultarea cu dl
sindromul Marfan
Acest sindrom este moștenit într-o autosomal dominanta, deci, dacă unul dintre părinți este bolnav, probabil pentru a trece boala puilor este de 50%. Gena care cauzeaza sindromul Marfan, este evidențiată. Aceasta gena fibinei localizată pe cromozomul 15. În cazul în care nu au fost identificate mutatii, poate ajuta la analiza grupurilor de legătură, în cazul în care au rude care suferă de sindromul Marfan. Ecografia prenatală a fătului nu va ajuta prea mult, deoarece cele mai multe dintre manifestările clinice nu sunt în mod necesar să apară la făt și nou-născut. Părinții bolnavi care doresc să discute posibilitatea de diagnostic prenatal, ar trebui să se concentreze pe consiliere genetica, preferabil inainte de sarcina, pentru a fi în măsură să ia în considerare și să efectueze teste genetice.
Sindromul alungit intervalul QT (Romano-Ward)
Această stare, la care leșinul însoțit intervalul QT alungit (în absența surditate - vezi sindromul Jervell, Lange-Nielsen.), Autozomal dominant Mostenire cu un risc de 50% de transmitere la descendenți. Teoretic, diagnosticul prenatal este posibil, utilizând electrocardiografie fetale cu o atenție sporită analizei T. dinți În practică, cele mai multe familii prefera nou-nascuti ECG cu terapia medicală și chirurgicală adecvată. Există o eterogenitate genetică semnificativă. unele gene sunt canale ionice au fost găsite, care pot provoca modificări în această tulburare.
Sindromul Jervell-Lange-Nielsen
Semnele acestui sindrom sunt prelungirea intervalului QT, ceea ce duce la stări de leșin, însoțită de o anomalie congenitală sau dobândită în copilarie severă pierdere de auz neurosenzorială. Această din urmă caracteristică vă permite să facă un diagnostic diferențial cu sindromul Romano-Ward. Pentru că descrie o încălcare a moștenit autosomal recesiv, riscul descendenților este scăzut.
stenoza aortica supravalvulara
Această anomalie poate apare sporadic, dar descrie o familie în care este moștenit autozomal dominant, cu diferite grade de exprimare. De multe ori a constatat stenoza pulmonară asociată. Datorită variabilității manifestărilor, chiar și în cadrul aceleiași familii este dificil de a evalua riscul pentru puii de parinti bolnavi. Pentru a stabili dacă este cazul sporadic și este asociată cu o genă dominantă, poate fi necesară examinarea cardiologice altor rude. Dacă aveți rude cu defecte cardiace relevante, riscul de urmași va fi de 50%.
Diagnosticul prenatal folosind ecocardiografia fetală este dificilă, dar ajută pentru a exclude alte defecte cardiace congenitale. În cele mai multe cazuri, diagnosticul poate fi efectuată după naștere, folosind tehnici chirurgicale adecvate și să efectueze corecție.
stenoza aortica supravalvulara si stenoza pulmonara prezent împreună cu alte anomalii cu sindrom Williams, care este o încălcare a caracteristice caracteristicile faciale grosier, întârzierea creșterii, probleme de comportament, giperkalydiemiey și sensibilitate crescută la sunete (giperakuzis). Cele mai multe cazuri de sindrom Williams este însoțit de o deleție submicroscopic de elastina genei în brațul lung al cromozomului 7. laborator Majoritatea genetice ofera un test special cromozom folosind hibridizare fluorescenta in situ. Sindromul Williams este cauzată de obicei ca o nouă ștergere și nu se repetă în timpul sarcinii urmatoare.
Sindromul Ellis-van Creveld
Sindromul Ellisavan Creveld este cunoscut și sub numele de displazie hondroektodermalnaya. semne caracteristice sunt polidactilie, nanism cu membre scurte, unghiilor displazie si boli cardiace congenitale, de obicei sub forma unui mare defect septal atrial.
Această condiție este moștenită într-un autozomal recesiva, așa că, dacă aveți un copil bolnav în următoarele sarcini cu risc este de 25%. Dar riscul pentru pacient descendenților inferior.
Exista doua mutatii genetice care sunt caracteristice sindromului Ellisavan Creveld, care sunt situate lângă brațul scurt al cromozomului 4.
sindromul Kartagener
Sindromul Kartagener caracterizat bronșiectazii, sinuzita recurentă, dekstrakardiey cu alte defecte ale inimii sau fără alte caracteristici și inversus parțială sau completă situs. Pentru diagnosticul poate necesita o morfologie de microscopie electronică a microvililor la care arată o scădere a numărului de dineinovyh interne și externe mânere. În general, statul este considerat ca un autozomal recesiva cu un risc scăzut pentru puii. Există trei potențial locusului genei, dar pentru populația europeană, cel mai probabil constituie o încălcare a genei dynein localizată pe cromozomul 9.
Video: medic-genetician în St. Petersburg
anomalii cromozomiale
anomalii cromozomiale de multe ori duce la încălcări ale inimii, malformații congenitale și alte dificultăți de învățare. Prin urmare, orice copil sau adult cu un defect cardiace congenitale, însoțite de dismorfice și dificultăți de învățare, trebuie să fie testat cariotip pentru a exclude anomaliile cromozomiale. Există combinații de caracteristici care sunt ușor de recunoscut. De exemplu, defectul septal atrioventricular adesea în sindromul Down (trisomia cromozomului 21), iar coarctație aortică este caracteristic fetele cu sindromul Turner (45H).
In prezent, pentru a identifica anomalii care nu se găsesc în analiza standard de cromozomi, există o serie de teste genetice folosind tehnici moleculare. fluorescenta metoda de hibridizare in situ nou detectează apărut cu deleții ale sindromului cromozomului si sindromul Williams Di Giorgi, atunci când există un microdeletion pe cromozomul 22. Tehnici mai noi, folosind ADN mikrostrelok sau „gena chips-uri“ pot oferi rezultate mai precise în analiza cromozomilor.
Anomaliile 22q cromozomiale au fost descrise clinic in diferite moduri, înainte de a fost detectată ștergerea. termenii „sindromul velokardiofatsialny“ Prin urmare, în literatura de specialitate, sau „sindromul Shprinttsena“. Trăsăturile caracteristice ale sindromului cromozomului 22q deleție sau di Giorgio sunt anomalii cardiace, în special defecte septale ventriculare și tetralogie Fallot combinate cu o despicătură palatului sau alte anomalii, trasaturi faciale caracteristice, tulburări mentale prost definite, hipoparatiroidismul și imunodeficiență. În acest caz, pot exista probleme psihice, inclusiv schizofrenie. In majoritatea cazurilor, di sindromul Giorgio apare sporadic, dar poate fi mostenita autozomal dominant și fenotipul - variază. De exemplu, părinții cu tulburări ușoare, cum ar fi palatoschizisului și septal ventricular copilului defect poate avea dificultăți de învățare și boli cardiace congenitale severe. 22q microdeletions cromozomiale au fost găsite în unele familii cu un timp aparent de moștenire dominant defecte cardiace izolate.
Bolile cardiace congenitale
Bolile cardiace congenitale apare la o frecvență de 0,5-1% din numărul total de nașteri. In cele mai multe cazuri (circa 90%), etiologia este necunoscută și sunt considerate ca fiind rezultatul moștenirii poligenică sau multifactorială. Numai aproximativ 3% din cazuri sunt mostenite conform legilor lui Mendel. În alte cazuri, cauza sunt anomalii cromozomiale și influențele de mediu.
În cazul în care cauza inimii mamei bolii este impactul mediului extern, riscul bolii la puii ei este mai mic decât riscul de moștenire poligenică. Un exemplu este rujeolă, care, de la introducerea practicii de imunizare în masă este rareori o cauza de persistente și persistența canalului arterial alte malformații congenitale ale inimii, dar poate fi factorul predominant în grupele de varsta.
Consilierea genetica este adesea necesar pacienții care au deja un copil cu boli cardiace congenitale. Cu toate acestea, cu îmbunătățirea în ultimii ani de cunoștințe și tratamentul chirurgical al multor pacienți cu vârsta congenitală ajunge la fertilă și au nevoie de consiliere cu privire la riscul de boli de inima urmașilor lor. au fost efectuate numeroase studii, va furniza informații cu privire la riscul de urmași în funcție de sexul pacientului și speciile de bază de viciu, pe care le are de suferit. Cu toate acestea, numărul celor care participă la aceste studii de pacienți este mică și nu pot fi reprezentative, iar numerele obținute de diferiți autori variază foarte mult. Se pare că, cu cât riscul pentru puii de pacienți de sex feminin decât pacienții de sex masculin din motive care nu sunt bine înțelese. Dacă există o boală, este doar jumătate defectul aspect este identic cu cel la dispoziția părintelui bolnav, iar acest lucru trebuie să fie luate în considerare în consilierea genetică. Părinții trebuie să știe că un copil poate fi incompatibil cu viața sau un defect incurabil, iar prognosticul ar trebui să fie discutate în momentul detectării problemelor.
hipertensiune pulmonară
hipertensiune pulmonară primară apare de obicei sporadic, mai ales la femei, dar descrie unele familii cu transmitere autozomal dominantă. S-a constatat că cauza este PLG BMPR2 mutatie cromozomul 2.
tulburări autoimune
Cele mai multe dintre ele sunt moștenite ca un singur tulburari genetice, dar fac parte din familie. Ele pot avea un efect direct asupra fătului, de exemplu, în cazul lupus eritematos sistemic (LES). Acesta din urmă este o boală autoimună care apare predominant la femei. Implicarea cardiacă este observată în 25% din cazuri, sub formă de pericardic cu sau fără efuziune. Simptome cardiace nu prevalează în mod necesar, ca boala este un multisistem, dar pacienții de sex feminin pot avea probleme obstetricale grave, inclusiv recurente avort, nastere prematura, si agravarea simptomelor bolii de bază în timpul sarcinii. Offspring femeilor cu posibile bloc cardiac complet care necesita terapie de intretinere la nou-născuți-perioada degenerării. Se pare că problema este un rezultat al trecerii de autoanticorpi traversează placenta.
cardiomiopatie
Cardiomiopatia hipertrofica este adesea moștenită în mod autosomal dominant. Boala începe de obicei la varsta adulta tineri decat in copilarie. pot să apară aritmia. Diagnosticul precoce contribuie la un tratament eficient. Acesta a identificat o serie de gene ale căror modificări conduc la cardiomiopatie, inclusiv inima genei de lanț greu (3-miozină genei troponinei T si gena -tropomiozina. Ecocardiografie fetus nu va recunoaște cardiomiopatie care începe la maturitate, iar diagnosticul prenatal depinde de identificarea corespunzătoare o mutatie in familie. se recomandă direcția pacienților la centrele specializate.
Idiopatica cardiomiopatie dilatativă este, de asemenea, un grup eterogen de tulburări, dar uneori poate avea caracteristici de boli ereditare. In cele mai multe familii, autozomal dominant, deși este descris într-o familie cu modul recesiv sau X-linked transmiteri autosomale. Variabilitatea expresie poate provoca dificultăți în consiliere. Aceasta ajută la clarificarea tipului de studiu moștenire de istorie de familie, dar în familii mici, pentru a detecta bolile ascunse mai bine oferi screening-ul ecocardiografic rudele de gradul întâi.
Defecte in arterele coronare si infarct miocardic
boala arterelor coronare poate fi atat genetica si a apărut sub influența mediului înconjurător. Acesta este rareori considerată din cauza riscului pentru urmași. Cu toate acestea, hipercolesterolemie familiala este o tulburare autosomal dominanta, care este asociat cu 10-20% din cazurile de boala precoce coronariene a avut loc, iar în cazul în care este diagnosticată în părinți, este mai bine să efectueze testarea copiilor, pentru a fi în măsură să ia măsuri preventive. In timp ce riscul unei astfel de gene moștenire familii este de 50%, un risc mai mic de boli de inima din cauza o varietate de factori suplimentari. Principalul defect in hipercolesterolemie familiala este lipsa de receptori pentru lipoproteine cu densitate scăzută, iar gena este localizată pe cromozomul 19. In multe cazuri, mutatii au fost identificate și măsuri adecvate pentru screening-ul în unele familii.
 Cauzele anomaliilor fetale. Riscul de apariție a malformațiilor fetale
Cauzele anomaliilor fetale. Riscul de apariție a malformațiilor fetale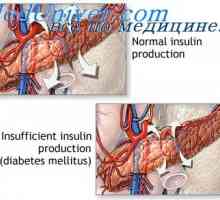 Alele dominante și recesive de cromozomi. dominantele autozomal
Alele dominante și recesive de cromozomi. dominantele autozomal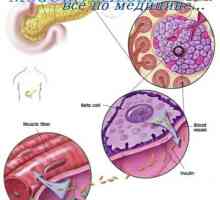 Moștenire autosomal recesivă. moștenire legată X-
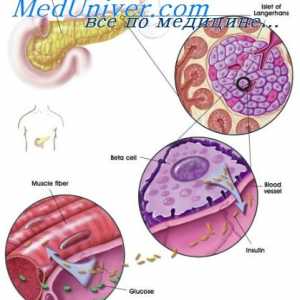
Moștenire autosomal recesivă. moștenire legată X- Cauza diabetului poate fi modificari genetice
Cauza diabetului poate fi modificari genetice Anomalii ale organelor genitale feminine. Sindroame Kaufman-mac-Cusick și…
Anomalii ale organelor genitale feminine. Sindroame Kaufman-mac-Cusick și… Diagnosticul genetic preimplantare (PGD). Indicații și posibilități
Diagnosticul genetic preimplantare (PGD). Indicații și posibilități Genomului uman. O gena o proteină adevărat dreapta?
Genomului uman. O gena o proteină adevărat dreapta?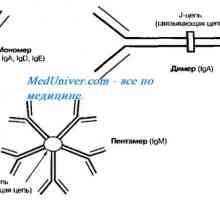 Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation
Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation- Gene noi Descoperit responsabile pentru cancerul de san
- Sindromul Peutz-Jeghers cauze, simptome si diagnosticul de sindrom Peutz-Jeghers
- Consilierea genetică în narcologie
- Genetica de consultare în timpul sarcinii
- Screening-ul în timpul sarcinii, screening genetic al nou-nascuti
- Care sunt bolile genetice
- Vărul meu, sindromul Down, și eu sunt îngrijorat de faptul că aceste anomalii cromozomiale vor fi…
 Riscul de a avea un copil cu malformații, studii genetice
Riscul de a avea un copil cu malformații, studii genetice Timing diagnosticul prenatal al bolilor genetice.
Timing diagnosticul prenatal al bolilor genetice. Gene boala fetale. Diagnosticul prenatal al bolilor genetice fetale.
Gene boala fetale. Diagnosticul prenatal al bolilor genetice fetale. Diagnosticul indirect al bolilor genetice. Acuratețea diagnosticului molecular al posibilelor surse…
Diagnosticul indirect al bolilor genetice. Acuratețea diagnosticului molecular al posibilelor surse…- Mamiferele sunt genetic mai asemănătoare cu părinții lor
 Sindroame monogenice mvpr
Sindroame monogenice mvpr
 Moștenire autosomal recesivă. moștenire legată X-
Moștenire autosomal recesivă. moștenire legată X- Diagnosticul indirect al bolilor genetice. Acuratețea diagnosticului molecular al posibilelor surse…
Diagnosticul indirect al bolilor genetice. Acuratețea diagnosticului molecular al posibilelor surse… Sindroame monogenice mvpr
Sindroame monogenice mvpr Diagnosticul genetic preimplantare (PGD). Indicații și posibilități
Diagnosticul genetic preimplantare (PGD). Indicații și posibilități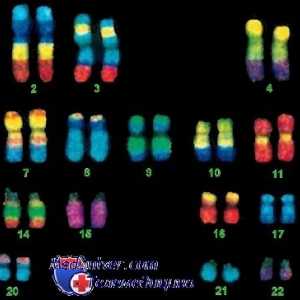 Genomului uman. O gena o proteină adevărat dreapta?
Genomului uman. O gena o proteină adevărat dreapta? Timing diagnosticul prenatal al bolilor genetice.
Timing diagnosticul prenatal al bolilor genetice.