Sindroame eponime: tratament

În acest articol ne uităm la principalele caracteristici ale sindroamelor omonim si tratamentul lor.
Aaza sindrom. Triada clinică a anemiei congenitale, tripla falangă a degetului mare și VSD. Etiologia este necunoscuta.
Sindromul Alfidi. Hipertensiune arterială, ca urmare a ocluzie a trunchiului celiac, ceea ce duce la fluxul de by-pass gUihă din artera renală dreaptă. descrisă inițial ca sindrom renal viscerală fura.
sindromul Andersen. Triada constând din paralizie periodică, tahiaritmii ventriculare si semne dysmorphia (hypertelorism, micrognathia, urechi set joase, palatoschizis, statura scurt, scolioză, syndactyly și clinodactyly). paralizie paroxistice pot apărea atunci când hiper-, hipo- sau normokalemia. Asociate cu mutații ale genei KCNJ2 care codifica canalele de potasiu rapid.
sindromul Barlow. forma PMC Familial, care, uneori, este moștenită ca o trăsătură autozomal dominantă. Genetic sindrom eterogen, caracterizat printr-un prolaps de una sau ambele valve în valvulelor atriul stâng în timpul sistolei. Auscultatie asculta srednesistolichesky clic și zgomot de întârziere sau pansystolic. 20% sunt asimptomatice. Femeile suferă în 2 ori mai des.
Sindromul Barth. gena Mutația TAZ localizată pe cromozomul X. Aceasta duce la cardiomiopatie dilatativă, miopatie, o creștere scăzută și neutropenie. Excreția 3 metilglutakonikovoy excretie acidă este aproape întotdeauna observate.
Sindromul Beemer malformații letale. rezultat fatal congenitale dublarea producției a ventriculului drept cu hidrocefalie, os de etanșare, trombocitopenie și dezvoltarea anormală a nasului.
Sindromul Buyo. Numele febră reumatică de autor. Buyo a fost primul care a subliniat importanța implicării în procesul patologic al inimii în faza acută a articular febră reumatică.
Boala-Pringle Burnevilya. inima hamartom și rinichi, în combinație cu epilepsie, retard mental, hamartoame corticale (scleroza tuberoasă) și adenom sebaceu. Pot să apară chisturi renale sau tumori.
Sindromul Bradburi-Eggleston. tulburare vegetativă idiopatic, caracterizată prin hipotensiune arterială ortostatică, cu manifestări frecvente de tulburări ale funcției intestinului, vezicii urinare, termoreglare și funcția sexuală.
Sindromul Brugada. Una dintre principalele cauze ale morții subite în rândul tinerilor, în absența unei boli cardiace organice, împotriva mutație genetică SCN5A. Perturbat canal de sodiu, ceea ce duce la apariția și menținerea aritmii ventriculare. Clinic manifest BPNPG, supradenivelarea segmentului ST în derivațiile V1-V3 și moarte subită sau sincopa. fenotip clinic poate fi detectat aymalina programare sau procainamidă. Singurul tratament eficient - implantarea unui defibrilator cardioverter.
Sindromul Carney. De asemenea, cunoscut sub numele de complex Carney. Această combinație de mixom cu mixom atrial în alte locuri, de exemplu sân, sau piele, si hiperactivitate pigmentare pestrițe glandelor endocrine, cum ar fi pituitare sau tumora testiculara. Provocate de o mutatie in cromozomul genei PRKAR1A 17. Sindromul se manifestă în vârstă de aproximativ 30 de ani, în majoritatea cazurilor, mixom bilaterale, care, spre deosebire de cazurile sporadice vor reapar după îndepărtarea.
sindromul DiGeorge. Tulburarea care rezultă din deleția genei TVH1 pe cromozomul 22q11.2, conducând la hipoplazie paratiroidian (și hipocalcemie), hipoplazia timusului (și niveluri scăzute ale limfocitelor T), anomalii ale tractului scurgere de sânge ale inimii, inclusiv tetradă Phaplu, malformație vasculară coloana vertebrală , întreruperea arcului aortic, dekstrapozitsiyu aortica, a adaugat artera subclavie. Pacientii cu acest sindrom sunt de obicei prezente micrognație, urechi set-joase, triunghi nasolabial scurt și o gură mică.
sindromul Shprinttsena De asemenea, a provocat o deranjat în aceeași genă.
Sindromul Dressler lui. Pericardita asociat cu infarct miocardic, în general, are loc după o săptămână după începerea atac de cord, dar pot apărea și câteva luni mai târziu. Sugerează origine autoimună din cauza întârzierii dezvoltării sindromului, prezența anticorpilor împotriva cardiomiocite, prezența limfocitelor modificate și activarea complementului, frecvent recidiva pleurită asociată și revărsat pleural, precum și sensibilitatea la NSAIDS și glucocorticoizi pot fi frecare zgomot pericard, febră, efuziunea pericardica si anomalii pleurale intervagyuv PR, ST, T schimbare val, care este sugestiv de pericardită.
distrofie musculară Duchenne. Concatenat cu tulburări legate-X, în care nu există nici o proteină particulară. Există o slăbiciune pronunțată a mușchilor scheletici, care pot masca cardiomiopatie dilatativă. Există o tendință de a fibroza si peretele posterolateral zadnebazalnoy a ventriculului stâng. aritmiile supraventriculare sunt mai frecvente decât fibrilație ventriculară și bloc atrioventricular, care apar ca fibroza cardiacă progresează.
anomalie Ebstein. Malformație, în care clapele observate de atașare patologice ale valvei tricuspide care conduce la deplasarea ventilului în jos. Prin urmare, o parte a ventriculului drept este situat între inelul atrioventricular și plasați atașamentul supapei, astfel încât partea proximală a ventriculului drept este de fapt atrium și cavitatea ventriculului drept este redusă. displazia Urmărirea a valvei tricuspide. Severitatea stării asociate cu stenoza sau atrezie pulmonara si cu ASD si VSD.
Sindromul Eisenmenger. Defect cu hipertensiune pulmonara si cianoza. Aceasta se produce atunci când presiunea în jumătatea dreaptă a inimii devine mai mare decât stânga, și există o descărcare de sânge venos în patul arterial. Acesta a fost descris pentru prima dată în 1897 de la un bărbat în vârstă de 32 de ani, cu un DSV.
Sindromul Ellis van Creveld (Displazia Hondroektodermalnaya). boala autozomal recesivă caracterizată prin statura scurt cauzate de displazie metafizară, polidactilie, unghiile displazia si dinti, si de obicei ASD. Coarctație de aortă, camere hipoplastic stang si artera spate descendent apare în 20% din cazuri.
distrofie musculară, Emery-Dreifuss. Clinic se manifestă prin triada constând din contracții timpurii ale cotului, tendonul lui Ahile și mușchii gâtului posterior, miopatie progresivă a musculaturii scheletice, și manifestări cardiace. În primul rând, acestea includ bradicardia sinusală, AF și flutter atrial, bloc atrioventricular apoi apar foarte constant VT si fibrilatie ventriculara. Se poate întâmpla, de asemenea, stop cardiac. De multe ori moarte subită înainte de vârsta de 50 de ani. Boala este legata de cromozomul X. Este legat de o gena care este responsabil pentru care codifica proteina de membrană nucleară numită emerin.
Tetralogie Fallot. Combinația de stenoza artera pulmonara, VSD, aorta dekstrapozitsii și hipertrofie ventriculară dreaptă care cauzează cianoză în tetralogia nou-născut Fallot este de 10% din toate defectele congenitale si este mai frecventa la barbati, inclusiv stenoza pulmonara, defect septal atrial si interventricular intacte pentad peregorodku- Fallot - tetralogie și ASD sau PFO.
ataxie Friedreich. Boala degenerativă a creierului si a maduvei spinarii caracterizate prin ataxie spinării, deformari osoase, disartrie, și cardiomiopatie. Adesea este lăsat hipertrofie ventriculară, precum și hipertrofia asimetrică a peretelui despărțitor. Cel puțin - cardiomiopatie dilatativă. Poate fi fibrilatie atriala. Este moștenită ca o trăsătură autozomal dominantă, cu mutația identificată ca fiind instabil GAA trinucleotide (guanina-adenin-adenina), detectate în primul intron al genei frataxin pe cromozomul 9q13.
Boala ataxie Friedreich. Colapsul bruscă a venelor jugulare, care au fost în prealabil extinse la fiecare diastola datorită pericardului adezive. De asemenea, cunoscut sub numele de mediastinoperikardit sau semna Fridreyha.Sindrom Holt-Oram. tulburare autozomală dominantă, de asemenea, cunoscut sub numele de sindromul inimii mână, care interferează cu creșterea extremităților superioare, de obicei, asociat cu secundar ASD, dar, de asemenea, cu VSD, PMK, persistența canalului arterial. Deformările membrului superior poate fi greu de observat (de exemplu, trehfalangovy distally dispuse sau degetul mare) sau pronunțat (hipoplazie claviculei și focomelia).
Sindromul Heidi. Combinația de sângerare gastrointestinală și stenoza de valva aortica cu kaptsifikatsiey. Conform descrierii inițiale în 1958 Heidi, la sângerare au apărut din cauza dobândite von Willebrand tip boala 2a cauzata de o presiune ridicată în spațiul inelar al valvei aortice, ceea ce duce la sangerare șunturilor arteriovenoase in intestin. Sangerarea se oprește atunci când proteza valvei.
Sindromul Hurler. tulburare autozomal recesiva determinând acumularea de deficit mucopolizaharide a enzimei lizozomale alfa-biduronata. De asemenea, este definit ca mucopolysaccharidosis tip IH (MPSlhi). Caracteristicile clinice includ caracteristici grosiere faciale, opacizare a corneei, gepatosplenomegapiyu, ingrosarea pielii, retard mental si patologie cardiaca. Aceasta include cardiomiopatie restrictivă din cauza fibroelastosis endomiocardica, stenoza arterelor coronare, îngroșarea valvulelor supapă (stânga mai mult de dreapta) și eșec. Cele mai multe mor în primii zece ani. Sindromul Hunter (MPS II) are un debit mai lent. Sindromul gatului (MPS IS) este cel mai favorabil pentru Mucopolysaccharidosis.
Sindromul Jervell-Lange-Nielsen. zabolevaenie autozomal recesivă însoțită de surditate, cauzata de o mutatie in gena KVLQT1 sau gena KCNE1 responsabile pentru canalele de potasiu lente implicate în formarea unui potențial de acțiune. Ca urmare a intervalului QT este prelungit, există un risc de tip tahicardie ventriculară „piruetă“ și moarte subită cardiacă.
Sindromul Kartagener. Triada clinică de situs inversus, patologia sinusurilor frontale și încă cilia de epiteliu ciliat. Pacienții observate infecții respiratorii recurente, sinuzita, bronșiectazie, și infertilitate pot fi unele niveluri anosmie sau scăzute de IgA. Moștenirea este autozomal recesiva. Defectul este într-o genă care codifică o proteină dynein, care determină structura epiteliului ciliar. De asemenea, cunoscut sub numele de sindromul de Sievert lui.
boala Kawasaki. vasculita acuta care afecteaza copiii si se manifesta cu febra, limfadenopatie cervicala, conjunctivită bilaterală, eritem, sau padoney scalarea si picioare, anevrisme arterei coronare sau Ectazia. Aceasta poate duce la infarct miocardic și moarte subită. Etiologia este necunoscuta.
sindromul Kearns-Sayre. triadă clinică constând din bloc atrioventricular, retinopatie pigmentară și ophthalmoplegia externe cronică progresivă. Boala este cauzata de o deleție a mai multor gene mitocondriale. În cele mai multe cazuri, dannoezabolevanie este un fenomen izolat și nu este moștenit.
nervului optic Ereditar Neuropatia Leber. encephalomyopathies mitocondriale caracterizate prin pierderea nedureroasa a vederii la o vârstă fragedă. Aceasta poate fi însoțită de un interval scurt de PR și emoție prematură.
Lenegre boala-Lev. Este, de asemenea, cunoscut sub numele de tip familial bloc I atrioventricular progresiva. tulburare autozomală dominantă asociată cu cromozomul 19, care sunt semne de pachet blocada bloc de ramură, complexele QRS sunt largi, care poate progresa spre o blocadă deplină. Boala se caracterizeaza prin familiala blocadă progresiva tip II, astfel încât acestea din urmă sunt complexele ORS înguste. Există un proces degenerativ accelerat care afectează în primul rând pe conductivitatea.
Sindromul Loeffler. O forma rara endocardita, urmata de un nivel ridicat de eozinofile circulante. Cauza principală poate fi o infecție cauzată de helminths sau leucemie, dar în cele mai multe cazuri, cauza principala este necunoscuta. Ca o regulă, cu raze X de lumină difuză-nod black-out. Forma acuta caracterizata prin vasculita eozinofilică, ceea ce duce la extinderea cavitățile inimii, în timp ce forma cronica provoaca fibroza miocardului, care, la rândul său, duce la sindromul clinic cardiomiopatia restrictivă din cauza care a redus toleranța la efort, acolo stridor, hepatomegalie, bloc atrioventricular, eșec tromboza valvei mitrale și tricuspide.
Sindromul Lown-Ganong-Levine. Fenomenul ventriculare premature caracterizate prin PR interval scurt (mai mic de 120 ms) și durata unui complex QRS normale, urmat de paroxismul SVT, dar fără AF sau flutter atrial. La pacienții fără antecedente de tahicardie au crescut conducerii atrioventriculare. Lown, Ganong si Levin soobshili despre acest sindrom în 1952, deși prima dată sindrom a fost descris ca funcționar în 1938 nu a fost detectat nici un anomalii structurale, care ar fi o cauză a sindromului Lown-Ganong-Levine. Boala se poate produce fibre parauzlovyh intrasite datorate sau ocolind nodul atrioventricular. Cu toate acestea, majoritatea pacienților alte cauze de tahicardie paroxistică a fost detectată prin studiul electrofiziologic, cum ar fi AVURT și nu ocolesc nodul atrioventricular. De aceea, sindromul Lown-Ganong-Levine - un sindrom legat de doelektrofiziologicheskoy studii care descriu epoca tahicardia paroxistică interval scurt de PR, care poate fi la limita inferioară a valorilor normale (2-4% dintre adulți PR interval mai mic de 120 ms).
Sindromul Libman-Sacks. Manifestarea lupus eritematos sistemic al inimii care apare tarziu in boala si se gaseste in 50% dintre pacienți după moarte. Caracterizata leziuni verucoase sterile supapă clapete și coardele, incluzând fibrină, neutrofile, limfocite si histiocite. De obicei, valvele mitrala si aortica uimit, cu toate că, în cele mai multe cazuri, nu există simptome clinice. Este mai frecventa insuficienta valvulara decat stenoza. Astfel de leziuni pot apărea cu sindrom antifosfolipidic. Boala femeile mai sensibile.
Sindromul Lutembacher. Combinația de stenoza mitrala (congenitale sau dobândite) și defect septal atrial (congenitală sau iatrogenă), cu șunt de la stânga la dreapta. În cazul în care ASD este mare, hipertensiune pulmonară poate fi evitată din cauza dilatarea inimii drept.
Sindromul Marfan. Tulburarea care rezultă din mutații ale genei FBN1 pe cromozomul 15q21.1, fibrilina 1 codificare care formează microfibrile care formează o matrice extracelulară. Unele dintre trăsăturile caracteristice ale sindromului - creștere ridicată, cifoză, scolioză, deformare pâlnie piept de deplasare în sus a cristalinului ochiului, expansiunea meningelor, dezlipire de retină și multiple tulburări cardiace (prolaps mitrala si insuficienta, dilatarea sinusurilor, dilatarea radacinii aortice cu eșec și stratificarea riscului crescut, aritmii). Pacienții pot fi un risc crescut de endocardită secundare patologiei valvulare. 75% este moștenită într-un autozomal dominant, restul - sunt cazuri izolate.
Sindromul Morquio. Una dintre tulburările însoțite de acumularea de mucopolizaharide - mucopolizaharidoza tip IVB, un mod autosomal recesivă moștenită. Există două tipuri. Tipul A este cauzata de o deficienta de galactozamină-6-sulfatază, în timp ce tipul B este cauzată de deficiența de beta-galactozidaza. Caracteristicile clinice includ statura scurt, modificări scheletice și perturbarea articulațiilor, opacifierea corneei, gepatomegapiyu, aortica si insuficienta mitrala. Insuficienta cardiaca poate fi din cauza cardiomiopatie infiltrativ sau din cauza unor defecțiuni.
Sindromul Noonan. sindrom dismorfic caracterizat prin tulburări cardiace, statura scurt, urechi set-joase, hipertelorism, surditatea si diateza hemoragica. Aceasta boala este moștenită în mod dominant autosomal. Probleme cardiace includ stenoza valvei pulmonare. Acest sindrom este uneori numit „de sex masculin sindromul Turner,“ desi afecteaza ambele sexe si nu are aberații cromozomiale.
Sindromul lui Ortner. Initial descris ca compresia nervului recurent laringian avansat la stânga atriului când stenoza tral E cauzează răgușeală datorită pareza vocale ori. Acesta este uneori folosit pentru a descrie orice inimă benigne sau proces intratoracice care afectează nervul laringian întoarcere. Nervul din stânga este afectată mai des decât dreptul din cauza situației speciale în raport cu arcul aortic.
Angina (varianta) Prinzmetal. Ea apare ca rezultat al spasm al arterelor coronare si poate conduce la bloc atrioventricular și IM. Acestea pot fi prevenite prin luarea de blocante ale canalelor de calciu lent acțiune prelungită.
sindromul Romano-Ward. stare autozomal dominanta cauzata de o mutatie in genele cromozomi 3, 4, 7, 11 și 21 care codifică diferitele componente ale canalelor de sodiu și de potasiu. Intervalul QT este prelungit, există un risc ridicat de a dezvolta torsade de tip tahicardie de pointes, și moarte subită. El nu este însoțită de surditate și, astfel, diferă de sindromul Jervell-Lange-Nielsen.
Sindromul Shprinttsena. Boala este cauzata de o mutatie in gena TVH1, care este de asemenea responsabil pentru sindromul DiGeorge. Caracteristici speciale: - anomalii cardiace (de obicei, VSD), palatoschizis, retard mintal și feței tipic cu un nas proeminent, fante înguste ochi, și micrognatie. După cum bine cunoscut sub numele de sindromul palatine kardiofatsialny.
Sindromul Stokes-Adams. Sincopă induse de aritmie. De asemenea, cunoscut sub numele de sindrom, sindromul Morgagni Spence.
coreea Sydenham. Manifestările tardive ale febrei reumatice, datorită reacției inflamatorii cauzate de autoanticorpi în ganglionii bazali, nucleul caudat, iar după grupa O infecție streptococică incepe de obicei la 3 luni după infectare primară, simptomele pot persista timp de 2 săptămâni. Se caracterizează prin mișcări involuntare ale discoordination musculare și instabilitate emoțională.
Sindromul Taussig-Bing. malformații congenitale.
Sindromul Tietze lui. inflamație cartilajului Rib de etiologie necunoscută. cartilaj Caracterizata tumora care separa sindromul de alte tipuri de inflamatie cartilajele costale. Tumora poate rămâne după ameliorarea durerii. sub rezerva bărbați în anii treizeci lor timpurie, în principal ale bolii.
Sindromul Townes-Brox. stat autozomal dominant, care combină atrezie anală, patologie renală, superioare, nivelul membrelor inferioare și urechi, și, în unele cazuri, însoțite de defecte cardiace, inclusiv defect septal atrial si VSD.
sindromul Turner. Boala, care rezultă din absența unuia dintre cromozomul X, CW. Caracteristicile sale sunt coarctation aortă, statura scurt, absența caracteristicilor sexuale secundare, pterigion de col uterin, valgus a articulației cotului și edem limfatic. Boala este una dintre mutatii cromozomiale cele mai comune.
Sindromul Tvidlera. Nu este un eponim strict. Fenomenul unei defecțiuni stimulator cardiac permanent ca urmare a expunerii pacientului la generatorul de impulsuri.
Wenckebach inima. Descrierea a inimii, care este situat pe linia mediană, și are o dimensiune mai mică. Aceeași boală cunoscută sub numele de mesocardium sau „agățat inima.“
Wenckebach fenomen. Forma II grad bloc atrioventricular, caracterizat prin intervalul de alungire progresivă ECG PRA până excitație atrială nu mai este deținut în ventricule.
Sindromul Williams. Nadkpapanny stenoza aortica congenitala cu stenoza arterei pulmonare periferice, hipercalcemie, fata elf, incapacitatea de a auto-identificare, retard mental, strabism și anomalii dentare. Ventriculul stâng poate fi hipertrofiate si sinusurile pot fi dilatate. Mai mult, a arterei coronare pot fi extinse sau sertizate și accelerat procesul aterosclerotic. Acești pacienți prezintă un risc mai mare de endocardită și moarte subită. Există o transmisie autosomal dominanta, în cazul în care sindromul moștenit.
Wolff-Parkinson sindrom-White. Tahiaritmii, care apar ca o consecință a modului atrioventricular suplimentare. Proprietăți ECG: ritm sinusal, intervale de PR mai mică de 120 ms, complexul QRS este lărgit mai mult de 120 ms, datorită D-wave (preexcitație), care rezultă din cauza anterograd desfășurarea cale conductivă suplimentar. Pacienții pot fi un comportament instabil, prin acest mod, ceea ce duce la modificări ECG.
 Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus
Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale
Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale Sindromul Apert la făt. Diagnosticul și prognosticul sindromului Apert
Sindromul Apert la făt. Diagnosticul și prognosticul sindromului Apert Sindromul Holt Oram. sindromul Gidroletalny
Sindromul Holt Oram. sindromul Gidroletalny Mikrotsefalichesky nanism primar. Sindromul dezordonate-Lax
Mikrotsefalichesky nanism primar. Sindromul dezordonate-Lax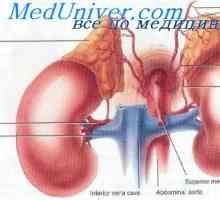 Sindromul Noonan într-un fat. oligohidramnios Sequence
Sindromul Noonan într-un fat. oligohidramnios Sequence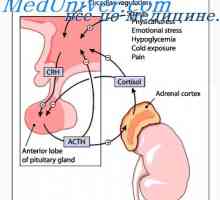 Sindromul Pfeiffer. Diagnosticul și prognosticul sindromului Pfeiffer
Sindromul Pfeiffer. Diagnosticul și prognosticul sindromului Pfeiffer Sindromul Pierre Robin. Displazia Septo-optică a fătului
Sindromul Pierre Robin. Displazia Septo-optică a fătului Anomalii ale organelor genitale feminine. Sindroame Kaufman-mac-Cusick și…
Anomalii ale organelor genitale feminine. Sindroame Kaufman-mac-Cusick și…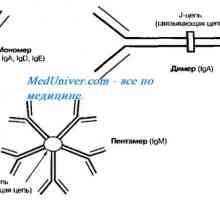 Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation
Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation Chist-stop-genital sindrom. Genetica de infertilitate masculină
Chist-stop-genital sindrom. Genetica de infertilitate masculină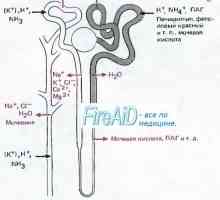 Sindromul Gitelman la copii. Diagnostic si tratament
Sindromul Gitelman la copii. Diagnostic si tratament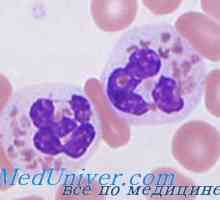 Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann
Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann Paroxysms Narcolepsia de somnolenta irezistibila cu dezvoltarea, în funcție de situația externă.…
Paroxysms Narcolepsia de somnolenta irezistibila cu dezvoltarea, în funcție de situația externă.…- Familie paralizie periodică (mioplegii familie paroxistică) boală ereditară caracterizată prin…
- Sănătate Enciclopedia, boli, medicamente, medic, farmacie, infecție, rezumate, sex, ginecologie,…
 Sindromul rezistenței la hormon tiroidian
Sindromul rezistenței la hormon tiroidian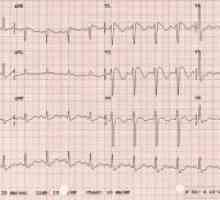 Sindromul Brugada: simptome, tratament, simptome, diagnostic
Sindromul Brugada: simptome, tratament, simptome, diagnostic Modificări în organul de vizibilitate în patologia congenitală a zonei faciale traumatice
Modificări în organul de vizibilitate în patologia congenitală a zonei faciale traumatice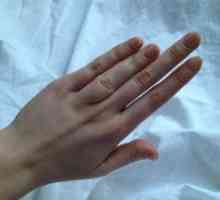 Degetul mare Brachydactyly: cauze, tratament, simptome, semne
Degetul mare Brachydactyly: cauze, tratament, simptome, semne Sindromul Noonan (sindromul Turner masculin)
Sindromul Noonan (sindromul Turner masculin)
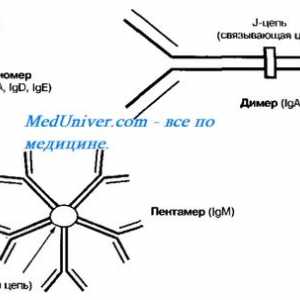 Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation
Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation Sindromul Noonan (sindromul Turner masculin)
Sindromul Noonan (sindromul Turner masculin) Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann
Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann Chist-stop-genital sindrom. Genetica de infertilitate masculină
Chist-stop-genital sindrom. Genetica de infertilitate masculină Sindromul Pierre Robin. Displazia Septo-optică a fătului
Sindromul Pierre Robin. Displazia Septo-optică a fătului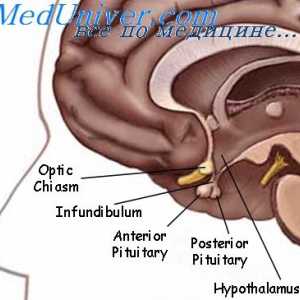 Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus
Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus Degetul mare Brachydactyly: cauze, tratament, simptome, semne
Degetul mare Brachydactyly: cauze, tratament, simptome, semne Copii Progeria: simptome, tratament, cauze
Copii Progeria: simptome, tratament, cauze Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale
Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale