Pseudohermafroditismului Barbat

conținut
- Rezistența la hcg testicular și lh
- Defecte în țesuturile țintă androgen-dependente
- Androgeni resistance aplicatii endpoint (defect al receptorului de androgen)
- Rezistență la androgenice la barbati, cu o structură normală a organelor genitale externe
- Defecte in metabolismul deficit de testosteron tkanyah- periferic 5a-reductaza-2 (psevdovaginalnaya perineoskrotalnaya hipospadias)
- Disgenetichnye 46, xy dpc (organe genitale, fără un anumit tip de structură datorită dysgenesis gonadică)
- Sindromul de regresie testiculara (sindromul disparut testicular, xy-agonadism, sindromul testicul rudimentar, anorchidism congenital)
- Sindromul de persistent mullerian (defecte de sinteză, secreție sau sensibilitate la antimyullerovomu hormonului)
- Produse chimice iatrogene
Când oamenii sunt pseudohermafroditismului gonade, care sunt ouăle, dar canalele genitale interne și a organelor genitale externe, sau ambele, iar celălalt nu este suficient de masculinized.
pseudohermafroditismului Male poate rezulta din deficit de testosteron din cauza: (1) diferențierea defectului testicular (dysgenesis testicular) - (2) tulburări de secreție de testosteron AMG- sau (3) reducerea sensibilității organelor țintă la testosteron si DHT AMG- sau (4) perturbarea conversia testosteronului in DHT.
Rezistența la hCG testicular și LH
diferențierea sexuală pentru bărbați depinde de producția de testosteron fetale de celule Leydig. În perioada critică a diferențierii sexuale masculine, secreția de testosteron de către celulele Leydig sub influența lui hCG placentare, iar apoi, în perioada de gestație - fetale Ki-pofizarnogo L G.
Cazurile de dezvoltarea normală a organelor genitale masculine la XY fetusi cu anencefalia, o absenta congenitala a glandei pituitare sau popituitarizmom hipotalamo-Gi sugerează că diferențierea sexuală masculină la om are loc indiferent de secreție fetale de gonadotropine.
Absența, hipoplazie sau amorțire celulelor Leydig la hCG-LH conduce la o deficienta in producerea de testosteron și, în consecință, pseudohermafroditismului masculin. Severitatea structurii genitale încălcări reflectă gradul de declin funcțional în producția de testosteron. Fenotip variază considerabil: forma cea mai pronunțată de încălcare a femeilor cu forme ușoare structura genitaliy- în care există masculi mikropenis- cu structura normala a organelor genitale externe și hipogonadism hipogonadotropic în timpul pubertata- în cele din urmă, în cele mai severe cazuri - testicule necoborat. Testiculele sunt, de obicei, de mici dimensiuni, după pubertate, au redus numărul de celule Leydig lipsesc sau, celulele Sertoli sunt normale, iar în tubii seminiferi nu sunt semne de spermatogeneză. S-a arătat că celulele absența, hipoplazie sau insensibilitate Leydig la unii pacienți pot fi o mutatie a genei care codifică receptorul hCG-LH, și a fost într-un model animal. Majoritatea pacienților cu un defect este notat structura de sex feminin a organelor genitale externe și vagin scurt se termină orbește. Există regresia completă a Mullerian protokov- unii pacienți chiar cu masculinizarea minimă a organelor genitale externe a relevat derivați conducte Wolffian. In post-pubertala pacienți au crescut concentrațiile bazale de gonadotropine, precum și răspunsul la stimularea cu GnRH conținut 17a-hidroxiprogesteron, androstendion și testosteron scăzut și rezultatele administrării de hCG într-un mic sau creștere zero, concentrațiile de testosteron sau precursori ai acestuia. Doi frați cu cele mai severe manifestări clinice ale acestui sindrom a fost descoperit de mutatie missense homozigotă a exonului 11-lea genei receptorilor G. L Această mutație conduce la substituția alaninei la prolină la al saselea domeniu codon transmembranar al formării receptor și receptorul LH non-funcțional. A fost descris mai târziu alte mutații de inactivare din familii care nu au legatura cu rezistenta la G. Un studiu efectuat la pacienți cu manifestări clinice și biochimice ale hipoplazie celulelor Leydig au aratat ca cauzele acestui sindrom geterogenny- ele pot fi ambele mutații în afara regiunii de codificare a receptorului hCG, LH și alte mutații gene specifice celulelor Leydig. Pacienții cu concentrații parțiale receptorilor LH determinarea inactivarea de steroizi sexuali pot fi destul de specific, dar receptorilor LH analiza genetica informativa.
Tratamentul depinde de vârsta de diagnostic și de gradul de masculinizare. Pentru pacienții cu o structură de sex feminin a organelor genitale externe este de obicei ales de sex feminin, cu toate că tuberculul sex (primordiurri falic) raspunde la testosteron. La pacienții cu tratament cu testosteron organelor genitale de tip predominant de sex masculin duce la dezvoltarea penisului și virilizare pacientului în timpul pubertății.
Defecte în țesuturile țintă androgen-dependente
Acesta a fost recent elucidat un mecanism complicat de acțiune a hormonilor steroizi la nivel celular. testosteron liber intra in celula tinta si este supus reducerii de 5a-DHT. DHT se leagă la un receptor de androgen intracelular induce modificări conformaționale care stimulează eliberarea de proteine de șoc termic, transport nuclear, dimerizarea și legarea la elementele ADN specifice pentru hormonii de împerechere. Acesta inițiază transcripția, translația și sinteza proteinelor, care se realizează un efect androgenic. efect androgenic insuficient în organul țintă și, în consecință, formarea de 46, XY DPC poate fi asociată cu transformarea complexă activitate anormală 5CX reductaza a unui receptor steroid, legarea receptorului de DHT, legarea complexului receptor-ligand cu un koregulyatorov ADN transcriere, eksportatsii transcriere sau traducere.
androgeni Resistance aplicatii endpoint (defect al receptorului de androgen)
Sindromul rezistentei complet (insensibilitate) de androgeni și variantele sale
Sindromul rezistentei complet (mort) androgen caracterizat cariotip 46, XY, testiculele bilaterialnymi, absența sau hipoplazia volfovyh conducte structura de tip femelă a organelor genitale externe cu labiile mici subdezvoltate și clitoris, care se încheie orbește vagin și derivații Mullerian absente sau rudimentare (33%) . În timpul pubertății se dezvolta caracteristicile sexuale feminine tipice secundare, dar nu se produce menarha.
cresterea parului pubian si axilar este, de obicei, o rară și o treime din pacienți complet absente. Pacientii de obicei peste înălțimea medie de sex feminin (pacienți înălțime medie 162,3 cm). Unii pacienți au o variantă a bolii, în care există o ușoară creștere a clitorisului. În timpul pubertății, acestea au, în plus față de dezvoltarea structurilor de sex feminin de san si, poate exista o virilizare moderat.
In timpul embriogenezei rezistentei previne pudendal androgeni masculinizarea și conductele de diferențiere volfovyh. Receptorii deteriorate pot reține o activitate reziduală și pentru a răspunde la concentrații locale mari de testosteron in vivo, ceea ce explica conductele derivatele de detecție volfovyh la unii pacienți cu rezistență aparentă androgen completă. Secreția de AMH de către celulele Sertoli ale fetale conduce la regresia Mullerian. Astfel, persoanele afectate se nasc cu organe genitale feminine și vaginul se termină orbește. În timpul pubertății, ca urmare a rezistentei crescute la secretia de androgeni de LH, urmat de concentrații crescute de testosteron si estradiol. Estradiolul este generată datorită conversiei periferice a androstendion și testosteron, și secretat direct de către testicule. Insensibilitate la androgeni, în combinație cu creșterea secreției testiculare de estradiol și conversia androgenilor la estrogeni are ca rezultat dezvoltarea caracteristicilor sexuale secundare în timpul pubertății. Timpul de creștere pubertar salt este aceeași ca și la fete sănătoase.
gena receptor androgenic este localizat pe cromozomul X Xq11 și între Xq13. Gena este compus din opt exonii sunt numerotate de la 1 la 8. exonul 1 codifică capătul amino terminal al proteinei receptorului androgen, și se crede că joacă un rol în reglarea transcripțională. Exonii 2 și 3 codifică segmentul de legare ADN al receptorului de proteină adrogennogo „zinc deget“. 5, o parte din exonul 4 al balamalei este numele și este implicat în legarea la nucleul celulei. Exonilor 5-8 sunt specifice carboxi terminal al receptorului androgen, care este un domeniu de legare androgen. 8 genă conține resturi de serină care pot fi fosforilate, 3 resturi de lizină, care pot fi acetilate și două resturi de lizină, care pot fi sumoylirovany. Sunt descrise mai mult de 600 de mutatii in receptorilor androgeni.
La pacienții cu insensibilitate completă de androgeni poate fi eterogenitatea în legarea DHT la receptorul androgen.
Descrisă pacientii receptor-negativ și receptor pozitiv cu receptor astfel de defecte de calitate ca termolabilității, instabilitatea și perturbarea capacității de legare, precum și pacienți cu aproape legarea normală.
Analiza androgen luminii varsat gena receptor pe baza eterogenității funcției receptorului marcat prin studiul pacienților cu rezistență completă androgeni. Pacientii cu receptori negativ de insensibilitate formă androgen completă în general, au mutații punctiforme sau substituții în 5-8 exonii care codifică-legare androgen retseptora- domeniu în regiunea de legare a ligandului receptorului androgen se observă un număr mare de mutații missense.
Alte defecte genetice, cum ar fi deleții, mutații ale sitului splice donor și mutații punctiforme care produc codonii de terminare prematură, sunt mai puțin frecvente în acest grup de pacienți. Mutațiile primul exon de obicei duce la o insensibilitate completă la androgenam- aproape toate dintre ele conduc la un codon stop prematur, rezultând într-un receptor nefunctional defect. Mutațiile în al treilea exon (care codifica receptorul androgen segmentul de legare a ADN-ului) sunt asociate cu legarea receptorului androgenic ligandului normal, dar la imposibilitatea complexului receptor ligand pus în contact cu ADN-ul și de a iniția transcripția ARNm. Aceste mutații conduc la receptori insensibilitate complet androgeni. Fenotipul pacienților afectați de obicei corelate cu studiile in vitro, gena andro-receptor și activitatea de transcripție a receptorilor ligandului androgen.
Alți factori care joacă un rol în exemplele de realizare genotip-fenotip includ mozaicism somatic koregulyatornye proteine transcripțional și alte gene „modificator“. mutații somatice - mutații care apar după formarea zigotului - conduc la prezența atât a receptorului normale si mutant androgeni. Datorită prezenței receptorilor normale la acești pacienți în timpul perioadei de pubertate, virilizare poate avea loc chiar și în cazul în care un copil au avut un tablou clinic clar insensibilității complet androgeni.
Diagnosticul complet de androgeni insensibilitate poate fi suspectat pe baza caracteristicilor clinice. Inainte de pubertate, diagnosticul se poate presupune în fete fenotipice cu formațiuni asemănătoare testiculelor in canalul inghinal sau labiilor. Aproximativ 1% din femeile cu hernii inghinale sunt rezistente la androgeni. La vârsta post-pubertar, pacienții au amenoree primară, dezvoltarea normală a glandelor mamare și absența sau de creștere a părului pubian rare sau axilar. Examinarea manuală sau ultrasonografie a confirmat absența colului uterin si a uterului.
Formele complete și incomplete ale rezistenței androgen trebuie diferențiate de alte forme de 46, XY DPC care rezultă din deficit de androgeni sau 5 reductaza. În prezent nu există metode disponibile, rapide pentru determinarea sensibilității la androgeni, in vivo sau in vitro. Sugerează diagnosticul se poate baza pe simptome clinice, antecedente familiale și bazale crescute și concentrațiile de testosteron stimulate hCG la fondul concentrațiilor normale de DHT. La sugari cu insensibilitate completă androgeni se observă creșterea de LH și testosteron, și ei nu au observat concentrații de recuperare post-natale ale acestor hormoni, cum este cazul la copiii sănătoși și cei cu sensibilitate incomplete la androgeni.
Reducerea insuficientă a legării steroizi sexuali globulină, la primirea unui stanozola steroid anabolic a fost propusă ca un test biologic pentru rezistenta la androgeni. Cu toate acestea, există foarte puține dovezi care să susțină posibilitatea utilizării sale. In plus, unii pacienți cu insensibilitate androgeni partiala a rezultatelor testelor poate fi la fel ca și la indivizii sănătoși. De asemenea, sa propus utilizarea unei creșteri concentrațiile AMH ca un marker al insensibilitate androgeni si deficit de androgeni, cu toate acestea, hormonul pot fi crescute în alte forme de 46, XY DPC. De înaltă precizie de diagnosticare trebuie să utilizeze definiția capacității de legare a androgenilor și transactivării, dar este o metode de lungă, consumatoare de timp și nu sunt disponibile pe scară largă. Analiza androgen mutatiilor genei de receptor sunt disponibile în prezent în multe laboratoare comerciale.
La copiii mici, cu organele genitale nedeterminate, problema de determinare a sexului, care este destul de complicată, efectuăm cercetări clinice: introducerea de testosteron enantat în soluție de ulei intramuscular la fiecare 4 săptămâni, timp de 3 luni, aceasta contribuie la evaluarea sensibilitatea la androgeni și pentru a identifica oportunitățile de creștere în continuare a penisului. Când sensibilitatea androgen intactă după un astfel de tratament lungimea penisului creste de minimum 1 cm.
Tratamentul pacienților cu insensibilitate completă androgeni este de a confirma și consolida identitatea lor de gen feminin. Din cauza riscului crescut de neoplasme gonadelor demonstrat castrare - indiferent înainte sau după pubertate. În cazul mosaicism somatice mai justificată pentru a transporta gonadectomy pubertate, pentru a evita posibila virilizare la pubertate. La pacienții care au suferit orhiectomie la pubertate ar trebui să înceapă terapia de substituție cu estrogen. În cele mai multe cazuri, nu este necesară o intervenție chirurgicală de plastic vaginale. Dacă este necesar, puteți crește dilatarea vaginului cu un manechin de plastic.
} {Modul direkt4
Sindromul de rezistență parțială (mort) de androgeni și variantele sale (sindromul de insensibilitate androgen parțial)
La pacienții cu insensibilitate androgen parțial la o gamă largă de manifestări fenotipice observate în funcție de gradul de virilizare. În timpul nașterii organelor genitale externe pot varia de la vag pentru a pune capăt orbește vagin la mascul hipoplazica. Există o variabilitate masculinizarea de oameni afectați, chiar printre rude. derivați de tubulatură Mullerian sunt absente conducte derivatele și prezente volfovyh, dar acestea sunt de obicei hipoplazica. În timpul pubertății vine virilizare slabă, există pubian și creșterea părului axilar, de multe ori se dezvolta ginecomastie. Cel mai frecvent fenotipul în postpubertate - de sex masculin, cu hipospadias perineoskrotalnoy și ginecomastie. cresterea parului pubian si axilar este de obicei normala. Testiculele rămân de obicei dimensiuni mici și azoospermie observate ca urmare a subdezvoltării celulelor terminale. Ca un copil, concentrații crescute de LH și testosteron, ceea ce nu se întâmplă la pacienții cu insensibilitate androgeni complet. Dupa pubertate, precum și la pacienții cu insensibilitate androgeni complet, concentrații crescute de LH, FSH, testosteron si estradiol. Cu toate acestea, în ciuda concentrațiilor crescute de estradiol, severitatea feminizare a acestor pacienți este mai puțin în comparație cu modul în care este la insensibilitate androgeni plin.
Investigarea receptorilor androgeni de la acești pacienți, de obicei, vă permite să vedeți anomalii calitative sau cantitative ale legării de androgeni. Evident, mutațiile care conduc la o reducere parțială a acțiunii androgenilor, va cauza virilizare incomplete. După cum sa menționat mai sus, cea mai bună corelare cu fenotip are un grad de afectare a activității de transcripție a receptorului-ligand androgen. O gamă largă de mutații diferite poate duce la același fenotip ca și mutația specifică poate fi asociat cu un alt fenotip la pacientii afectati. În general, o parțială (incompletă) androgeni insensibilitate conduc adesea la mutații punctiforme, rezultând în substituția aminoacizilor are loc. Deeb și colab. Am studiat 111 de pacienti cu insensibilitate androgeni partiala. Dintre cei 111 de pacienti, a caror ADN a fost examinat, mutații au fost identificate în doar 27 (24%). Majoritatea pacientilor cu mutatii gasit a existat o violare a legării DHT la receptorul en-drogennym. Cercetatorii au confirmat faptul că fenotipul este variabilă și foarte independent de genotip. Înainte de sexare finală în copilarie a verificat ca toti pacientii cu insensibilitate androgeni partiala este necesar pentru a efectua un tratament proces de testosteron, mai ales în cazul mosaicism somatice, pot fi prezente la receptorii androgeni. Datorită sensibilității androgen „parțială“ a acestor pacienți pot necesita doze mai mari de androgeni, în comparație cu convenționale, deoarece sa demonstrat că, în unele cazuri, acestea raspund la doze mari de testosteron. Dacă decideți să crească copilul în domeniul femeilor, ar trebui să fie efectuate înainte de pubertate gonadectomy, pentru a evita riscul posibil de virilizare la pubertate. Datele privind monitorizarea pe termen lung a pacienților cu insensibilitate androgeni partiala indica faptul ca multi pacienti sunt nemulțumiți de sexul lor, fie de sex masculin sau feminin în domeniu, acestea sunt readuse. Bouvettier și colab. Ei au observat că persoanele care au fost crescuți în domeniu bărbați, există adesea o reducere semnificativă în toate aspectele funcției sexuale.
rezistență la androgenice la barbati, cu o structură normală a organelor genitale externe
insensibilitate androgeni parțial descrisă la bărbați infertili cu fenotip normal de sex masculin, care de multe ori dezvolta ginecomastie. Unele dintre ele au concentrații normale de LH și testosteron, ceea ce le distinge de pacientii cu insensibilitate androgeni. Infertilitatea poate fi singura manifestare a insensibilitate androgeni în aceste normale în toate celelalte privințe bărbați. Într-una din familiile afectate au avut 5 masculi fenotipice cu ginecomastie si penis mic. Concentrațiile de testosteron au fost crescute și au relevat încălcări de calitate scăzută a ligandului de legare. Fertilitatea a fost documentată în 4 din acești 5 pacienți. Aceasta este cea mai usoara versiune de insensibilitate androgeni.
Defecte in metabolismul deficit de testosteron tkanyah- periferic 5a-reductaza-2 (psevdovaginalnaya perineoskrotalnaya hipospadias)
Violarea conversia testosteronului in DHT este cauza unui unic 46, XY PND. Fenotipic acești pacienți pot varia de la cei cu micropenis la pacienții cu hipospadias psevdovaginalnoy perineoskrotalnoy. La nastere, pacienții cu leziune cea mai severă observată penis mic cu hipospadias, coardă, scrot divizat și sinusul urogenital, deschidere între picioare. Prezent vagin se încheie orbește, care se deschide sau sinusul urogenital, sau în jurul uretrei, imediat după deschiderea uretral. Testiculele sunt canale inghinal sau labiilor. Structuri Mullerian sunt conducte Wolffian absente sunt bine diferențiate. În timpul pubertății se produce virilizatsiya- schimbat cu o voce aspră, creste masa musculara, penisul. scrot Split, devine pliat și pigmentat. Testiculele creștere și scădere a labioskrotalnye falduri, și poate începe spermatogenezei. Ginecomastia la acești pacienți nu se observă. Se poate observa lipsa acnee, parul de pe corp și chel patch-uri pe corp și membre. Caracteristica distinctivă a acestei forme de pseudohermafroditismului masculine este descrisă de identificare solicitare de schimbare cu masculul în timpul pubertății.
După instalarea pubertății la pacienții cu 5a-reductaza-2 deficiență au niveluri normale sau crescute de testosteron și concentrații ușor crescute de LH în sânge. Evident, lipsa 5a-reducere a testosteronului în DHT în fazele critice ale diferențierii sexuale masculine în timpul dezvoltării fetale duce la masculinizarea incompleta a sinusului urogenital și organelor genitale externe, structurile Wolffian dependente de testosteron se dezvolta normal. La pacienții cu rd hipospadias și / sau micropenis descrise forme parțiale sau ușoare ale 5a-reductaza. Trei frati de sex masculin din Suedia, care a avut un deficit de heterozigot compus 5C-reductaza-2, marcate hipospadias corectate în fază incipientă, iar două dintre ele au fost mai târziu de fertilitate.
deficit de reductaza 5C-2 este moștenită în mod recesiv autozomal și a observat heterogenitatea genetică a acestui defect enzimatic. Există două tipuri de persoane afectate: cei care nu au activitatea enzimei, și cele în care este definit, dar este instabilă. Două gene catalizează conversia testosteronului in DHT, iar acestea sunt numite de tip 1 și 2. Enzima de tip 1 nu este exprimat in fat, dar este exprimat în piele postnatal până la vârsta de 2-3 ani, iar apoi rămâne la un nivel scăzut până la pubertate. Tipul 1 gena nu a descris mutații. tip 2 izoenzima este o enzimă care se găsește în piele de fructe genitală, glandele accesorii masculine si prostata. Pacienții cu deficit de 5a-reductaza există o deficiență a enzimei pentru care pH-ul optim este de 5,5 (tip 2). Gena care codifică această enzimă conține 5 exoni și este localizată pe cromozomul 2, porțiunea p23. Descris de multe mutatii diferite, inclusiv ștergeri, nonsens, defecte de îmbinare și cele mai comune mutatie missense. Două treimi dintre pacienți sunt homozigoți pentru aceeași mutație, iar restul - Compoundat heterozigoții. Sa sugerat că virilizare semnificative, semne de pubertate, în ciuda lipsei de in utero, poate fi o consecință a expresiei și a funcției de tipul genei 1 la pubertate, ceea ce duce la producerea unei cantități de DHT în țesutul periferic, ceea ce este suficient pentru a induce o creștere a penisului și alte manifestări ale masculinizare.
Eșecul 5CX-reductaza-2 este de a fi suspectat în cazuri de pseudohermafroditismului de sex masculin, în care există un vagin orb, precum și la bărbați cu hipospadias și mikrofallosom. Diagnosticul este confirmat prin detectarea unui raport anormal de mare bazale și testosteron hCG stimulate și concentrațiile de DHT. Alte caracteristici, ceea ce confirmă diagnosticul, mai ales la nou-născuți, se referă au crescut cu 5 raportul (3 :. 5a metaboliții steroizi C21 și C19 în urină, în unele laboratoare pot examina nivelul de activitate al 5a-reductazei în cultură celulele genitale ale pielii și pentru a determina gradul de conversie a testosteronului etichetat introduse la DHT in vivo.
Deosebit de important este diagnosticarea precoce a acestei condiții. Având în vedere caracteristicile acestei boli, se arată identificarea în tratamentul masculin câmp DHT sau doze mari de testosteron necesare pentru marirea penisului inițiere. Corectarea hipospadias trebuie efectuate in copilarie sau copilarie. Pacienții care au fost diagnosticați mai târziu, care după pubertate există o identificare de sex feminin, metodele de selecție de tratament sunt organe genitale din plastic, orhiectomie preventivă și terapia de substituție cu estrogen.
Disgenetichnye 46, XY DPC (organe genitale, fără un anumit tip de structură datorită dysgenesis gonadică)
Formarea structurii de tip dublă a organelor genitale externe și interne și formarea sinusul urogenital poate rezulta din tulburări ale gonadogenesis testicular. gonadogenesis Violarea și, prin urmare, virilizare violare indicată la pacienții cu 45, X / 46, mosaicism XY, anomalii structurale Y cromozomi, și diferite forme de disgenezii gonadali. Pacienții cu 46, XY degenerență gonodală sunt clasificate în funcție de tipul de încălcare a diferențierii sexuale, cu toate acestea, este una dintre subgrupurile de pseudohermafroditismului de sex masculin. 46, XY disgenezii gonadelor, si este asociat cu o genă deleție mutație SPYna cromozomului Y, DAX1 gena duplicare (ANS) pe cromozomul X, Wnt4 duplicarea pe cromozomul 1 și ștergeri pe cromozomul 9r- (DMRT1,2) sau 10q-. În plus, disgenetichnym 46, XY DPC plumb mutație SOX9 (kampomelicheskaya displazie), DHCR7 (sindromul Smith-Lemli-Opitz) și XH2 (ATR-X Sindromul: talasemie X-linked, retard mintal, testiculele mici și tipul dual al structurii genitale ).
gena Due heterozigoti supresor de mutație antiNogo Wilms pseudohermafroditismului tumora disgenetichny mascul apare in asociere cu precoce manifestata prin boli degenerative renale si hipertensiune arteriala, precum si tumora Wilms (sindromul Denis-Drash) sau cu boli renale mai tarziu si risc crescut de gonadoblastom de gonad stivă (sindromul Fraser). Când sindrom mutatii Denys-Drash adesea găsit în exonul 9 al genei WT1. Fraser sindromul Cand mutatii apar in donor situs de splicing intronul 9, care duce la o relație inversă KTS + (lizina-treonina, serina) / izoformele proteinei KTB- WT1 în celulele țintă. La pacienții cu o deleție a genei WT1 are sindromul Warg și organele genitale sunt hipoplazia ambigue sau de sex masculin, inclusiv divizarea scrot, hipospadias si criptorhidie.
46, XY disgeneză gonadal mutații genetice asociate care codifică SF-1. In mod similar, șoarecii «knock-out», persoanele afectate sunt gonadele XY stivuibile cu diferențierea sexuală feminină și insuficiență suprarenală severă, care se manifestă în perioada neonatală. Cu toate acestea, o persoană în astfel de cazuri sunt găsite mutații heterozigote (prezente în doar o alelă a genei SF-1) și se menține secreția de gonadotrofine (în timp ce în «knockout» soareci mutatie homozigota a Sf-1 și gonadotropinei deficiență). mutația homozigotă a SF-1 a fost detectată cu fenotipul de mai sus, dar membrii familiei heterozigote chestionate au fost sanatosi. mutatie heterozigota a SF-1 a fost descrisă în cazul 46, XY degenerență gonodală cu functie normala a glandelor suprarenale. Aceste cazuri recente sugerează că diferențierea testiculară este un mod dependent de doză în comparație cu ontogeniei suprarenală. În 46, XX femeile cu mutatii observate insuficienta suprarenala geterogizotnymi si functia ovariana normala.
Sindromul de regresie testiculara (sindromul disparut testicular, XY-agonadism, sindromul testicul rudimentar, anorchidism congenital)
Încetarea funcției testiculare în perioadele critice ale diferențierii sexuale masculine conduce la o varietate de sindroame clinice, în funcție de momentul în care funcția de întrerupere a testiculelor. Pe de o parte, aceste spectru clinic heterogen de stări așa-numita XY-gonadale disgeneză - Pacientii 46, XY, a cărui funcție a încetat testiculare până la 8 săptămâni de gestație și a organelor genitale externe și interne formate prin care de tipul de sex feminin. Pe de altă parte - cei cu „anorchidism“ sau „ouă au dispărut“, ale căror testicule s-au lovit în etapele ulterioare de gestație. La acești pacienți, o diferențiere de sex masculin absolut normala a organelor genitale externe și interne, dar nici un tesut gonadale.
Diagnosticul Anorchidism trebuie să fie excluși toți oamenii cu criptorhidie. Un test util pentru evaluarea activității funcționale a celulelor Leydig este administrarea de hCG, 1000-2000 unități / m2 intramuscular la fiecare a doua zi timp de 2 săptămâni (un total de 7 injecții). Dacă celulele Leydig sunt prezente în prepuberi remarcat concentrațiile de testosteron crește de la mai puțin de 20 ng / dl (0,69 nmol / l) la mai mult de 200 ng / dl (6,9 nmol / l).
La copiii cu vârsta mai mică de 4 ani și după 10 de ani, un indicator sensibil al lipsei de țesut gonadale sunt concentrații crescute de FSH. Pentru a determina absența celulelor Sertoli și în consecință, este utilă definirea AMH anorchidism și concentrațiile inhibinei.
Pentru diagnosticul absenței feedback-ului din hipotalamus gonadelor și hipofizei poate fi utilizat cu un test prin administrarea intravenoasă a 100 mg de GnRH. Copiii cu agonadism remarcat o creștere mai pronunțată a concentrațiilor de LH și FSH, comparativ cu copiii cu funcție normală a gonadelor-pubertare pre. La pacienții cu concentrații ridicate de gonadotropine și testosteron lipsa de răspuns după administrarea de hCG este, de obicei, nu este posibil de a găsi țesut testicular în timpul procedurilor chirurgicale.
Sindromul de persistent Mullerian (defecte de sinteză, secreție sau sensibilitate la antimyullerovomu hormonului)
Acest sindrom, care se găsește numai la bărbați și este moștenită în mod autozomal recesivă, caracterizată prin prezența diferențierii sexuale masculine normale a organelor genitale externe, și în același timp păstrând derivații Mullerian. Motivele derivaților Mullerian de siguranță poate fi fie în încălcarea sintezei AMH de către celulele Sertoli, sau defecte ca raspuns la AMG organ țintă. Gena care codifică AMG, localizată pe cromozomul 19 mutatii cauzali sunt mai multe variante de gene AMG și gena care codifică AMG receptor. Indivizii cu mutații receptorului AMH stocate de structuri Mullerian, in ciuda niveluri normale sau ridicate de AMH în sânge. Tratamentul presupune eliminarea structurilor Mullerian.
produse chimice iatrogene
De-a lungul ultimilor 50 de ani a fost o creștere a frecvenței de încălcări ale dezvoltării și funcționării tractului urogenital la barbati. S-a emis ipoteza că o creștere a incidenței anomaliilor de reproducere observate la bărbați este asociată cu expunerea crescută la „estrogen“ în produse - atât de origine naturală și chimică - în timpul perioadei prenatale. S-a demonstrat că la rozătoare p, P '-DDE (dihlorodifenildih-loroetilen) - și metabolitul principal DDT constant - se leagă la receptorul androgen si inhiba functia de androgeni. Sunt necesare cercetari suplimentare pentru a evalua conținutul diferitelor substanțe chimice și hormonale în mediu, precum și riscurile de impactul lor asupra formării de anomalii ale sistemului de reproducere.
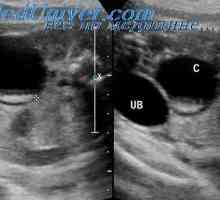 Criptorhidismul la făt. feminizarea testiculara
Criptorhidismul la făt. feminizarea testiculara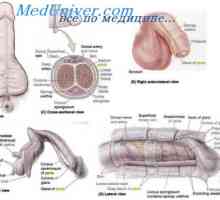 Metabolismul testosteron. funcţia de testosteron
Metabolismul testosteron. funcţia de testosteron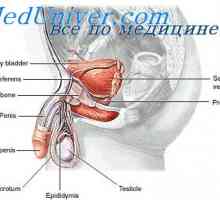 Testosteronul. Secreția de androgeni
Testosteronul. Secreția de androgeni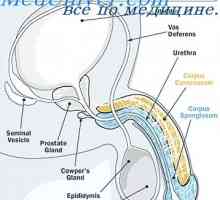 Suprimarea sintezei de hormoni masculini. Regulamentul spermatogenezei
Suprimarea sintezei de hormoni masculini. Regulamentul spermatogenezei Factorii psihologici ai activității sexuale. Gonadotropina corionică umană
Factorii psihologici ai activității sexuale. Gonadotropina corionică umană Încălcările funcțiilor prostatei. Hipogonadism la bărbați
Încălcările funcțiilor prostatei. Hipogonadism la bărbați Mutațiile gonadotropină receptori. Anomalii ale LH și FSH receptorilor
Mutațiile gonadotropină receptori. Anomalii ale LH și FSH receptorilor Structura testiculelor. Celulele Leydig și Sertoli
Structura testiculelor. Celulele Leydig și Sertoli Barbatii cu un nivel ridicat de imunitate atractive pentru femei
Barbatii cu un nivel ridicat de imunitate atractive pentru femei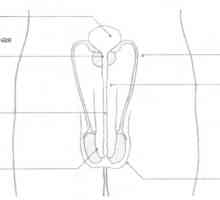 Glande și hormoni sexuali
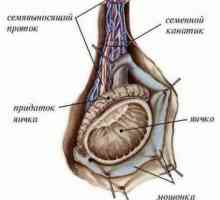
Glande și hormoni sexuali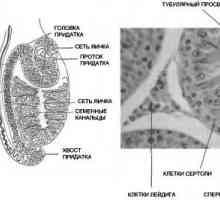 Anatomia și fiziologia sistemului reproductiv masculin
Anatomia și fiziologia sistemului reproductiv masculin- Tulburări congenitale ale differentsirovkizabolevaniya sexuale cauzate de anomalii cromozomiale.…
- Sănătate Enciclopedia, boli, medicamente, medic, farmacie, infecție, rezumate, sex, ginecologie,…
- Sănătate Enciclopedia, boli, medicamente, medic, farmacie, infecție, rezumate, sex, ginecologie,…
 Diferențierea testiculului și ovar
Diferențierea testiculului și ovar True hermaphroditism
True hermaphroditism Aplazie Leydig
Aplazie Leydig Anorchidism bilaterală (sindromul testiculelor disparut)
Anorchidism bilaterală (sindromul testiculelor disparut) Andropause (scăderea funcției celulelor Leydig)
Andropause (scăderea funcției celulelor Leydig) Micropenis, tratament
Micropenis, tratament Distrofie miotonica
Distrofie miotonica
 Factorii psihologici ai activității sexuale. Gonadotropina corionică umană
Factorii psihologici ai activității sexuale. Gonadotropina corionică umană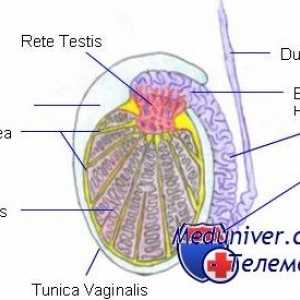 Structura testiculelor. Celulele Leydig și Sertoli
Structura testiculelor. Celulele Leydig și Sertoli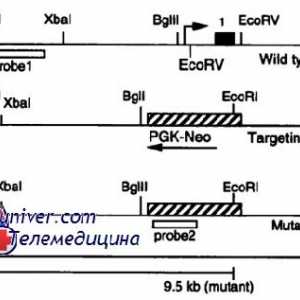 Mutațiile gonadotropină receptori. Anomalii ale LH și FSH receptorilor
Mutațiile gonadotropină receptori. Anomalii ale LH și FSH receptorilor Glande și hormoni sexuali
Glande și hormoni sexuali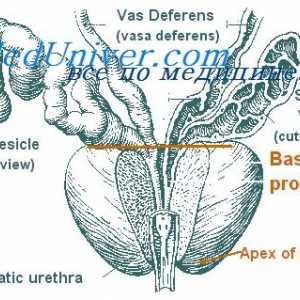 Încălcările funcțiilor prostatei. Hipogonadism la bărbați
Încălcările funcțiilor prostatei. Hipogonadism la bărbați True hermaphroditism
True hermaphroditism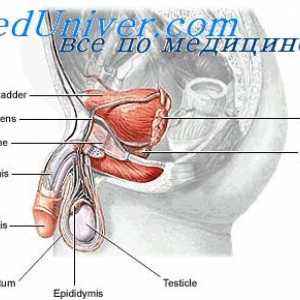 Testosteronul. Secreția de androgeni
Testosteronul. Secreția de androgeni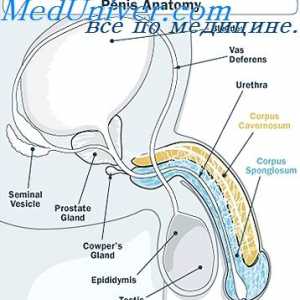 Suprimarea sintezei de hormoni masculini. Regulamentul spermatogenezei
Suprimarea sintezei de hormoni masculini. Regulamentul spermatogenezei