Sindroame genetice, assotsiirovannn cu feocromocitom și paraganglioma

conținut
Cele mai multe feocromocitoamele - sporadice, deși un număr semnificativ de aceste tumori sunt găsite mutații somatice similare embrionare (care stau la baza sindroame familiale).
Pe baza datelor din istoricul familial, înainte de crezut că doar 10% dintre pacienții cu astfel de tumori sunt parte din sindroame genetice. Cu toate acestea, odată cu introducerea metodelor de analiză genetică a arătat că 20-30% dintre pacienții cu feocromocitom și paraganglioma sunt purtători ai mutației germinale, determină dezvoltarea sindroamelor familiale. Astfel, aproximativ 9% dintre pacienții cu feocromocitom unilateral in care tumora pare mutatii sporadice detectate VHL (gena boala Von Hippel-Lindau). Prin urmare, analiza genetică se recomandă la toți pacienții cu feocromocitom și paraganglioma, mai ales atunci când paragangliomas extraadrenal, tumori multifocale, dacă simptomele la vârstă tânără (50 ani) sau în prezența tumorii în istoria familiei. Studiile genetice efectuate la pacienții cu alte manifestări sindroame ereditare, încercând să detecteze mutații RET proto-oncogena (sindromul II MEN) VHL gena (von boala Hippel-Lindau), NF-1 (neurofibromatoza) și gena LDH (SDGV și SDGD SDG).
Multiple neoplazie endocrina tip 2 (MEN II)
Baza sindromului autozomal dominant MAN II (cancer tiroidian medular, feocromocitom, și o serie de alte manifestări patologice) constă activarea pro-toonkogena mutație RET este localizată pe cromozomul 10, care codifică o tirozin kinază receptor transmembranar care este exprimat în țesuturi care provin din creasta neurală. Există două subtipuri ale acestui sindrom - MEN Pa (90%) și MEN lib (10%). Pentru pacienții cu sindrom MEN Pa caracterizate printr-o varietate de mutații missense care conduc la substituția unui aminoacid în domeniul extracelular al moleculelor de tirozin kinazei care sunt responsabile pentru homodimerization și activarea constitutiva. La pacienții cu sindrom MEN lib detectat mutația missense (codonului 918 din exonul 16) care modifică structura domeniului catalitic al tirozin kinazei intracelulare și, de asemenea, conduce la activarea constitutivă. In ambele subtipuri ale sindromului de obicei se dezvolta feocromocitom in paraganglioma extraadrenal nadpochechnikah- sunt rare. Hipertensiunea este de obicei caracterizat prin paroxistic peste. Fiecare mutație determină tipul de fenotip (vârsta la debutul și agresivitatea cancerului tiroidian medular).
activarea constitutiva a mutațiilor tirozin kinazei provoca doar câteva Exons RET. Ele afectează de obicei exonii 10, 11, 13, 14 și 15. De aceea, studiile genetice pot fi limitate numai la aceste exonilor. În cazul în care nu sunt detectate mutații, apoi în laboratoare speciale secvențiat rămase 15 exoni. Atunci când mutația în familie este cunoscut în prealabil, o analiza genetica a unui anumit pacient din familie a fost limitat la găsirea acestei mutații.
Cele mai multe cazuri de sindrom feocromocitom MEN II don datorită mutațiilor genei RET 634. Pacienții cu aceste mutații este necesar să se respecte și să examineze foarte atent pentru a identifica feocromocitom. Deoarece, în aceste cazuri, această tumoare se dezvoltă la o vârstă fragedă (aproximativ 5 ani), este necesar să se înceapă căutarea unui copil măsoare în mod regulat tensiunea arterială și determinarea nivelului de plasmă metanefrină.
Feocromocitomului sunt caracteristice pentru toate familiile cu sindromul MEN II, cu excepția asociată cu mutații ale genei RET la codonii 609, 768, și 891. val804met Pacienții cu astfel de mutații pot fi inspectate mai puțin frecvent.
Metastaze cu sindromul MEN feocromocitom II găsit în doar aproximativ 4% din cazuri, care este probabil asociat cu detectarea precoce a acestor tumori. feocromocitoamele bilaterale se găsesc aproape 70% dintre pacienți, dar la început mai devreme la un sondaj, cu atât mai des puteți găsi o tumoare unilaterală. După unilateral feocromocitom adrenal adrenalectomia în al doilea apare la aproximativ 50% dintre pacienți (vârsta medie 12). feocromocitom adrenal la sindromul MEN II secreta noradrenalina si epinefrina (si metanefrină metabolitul său). Conținutul de catecolamine în plasmă poate fi normal, dar nivelul metanefrină este de obicei crescut. Prin urmare, este cifra este cel mai bun marker al feocromocitoamele mici. În unele cazuri, cu toate acestea, feocromocitom poate fi detectată numai dacă o determinare separată a metanefrine, catecolamine și creatinină în urină de zi cu zi.
- MEN IIa (Sindromul Sipple). In acest sindrom, cancer tiroidian medular este detectat la 95-100% dintre pacienți, hiperparatiroidism (datorită hiperplazia glandelor paratiroide) - 35%, iar feocromocitom sau hiperplaziei medulosuprarenalei - 50% (variind de la 6% până la 100% în familii diferite ). Acești pacienți sunt adesea găsit ca lichenul plan si boala Hirschsprung. Feocromocitom apar de obicei la vârsta de mijloc, și nu este întotdeauna însoțită de hipertensiune arterială. RET Analiza proto-oncogena in familiile cu sindrom MEN Pa ar trebui să fie de până la 6 ani. Acest lucru vă permite să determine necesitatea tiroidectomie profilactică și de cercetare atentă cu scopul de a identifica feocromocitomul și hiperparatiroidism.
- MEN IIb. Acest sindrom include, de obicei, cancerul tiroidian medular, neuroame mucoase multiple (vizibile ochilor si a gurii), nervii corneene de îngroșare, aspectul marfanoidnuyu ganglionila-ROM-uri intestinului și feocromocitom sau hiperplazia medulosuprarenalei. cancer tiroidian medular apare mai agresiv si se dezvolta la o varsta mai devreme decat la barbati sindromul Pa. Posibilitatea de operatorii de transport mutatie de familie de RET proto-oncogene la pacient trebuie verificate imediat după detectarea unui feocromocitom. În identificarea unei astfel de mutatie este o tiroidectomie profilactic. Toate mass-media mutații adecvate necesită o supraveghere medicală atentă și examinare.
boala von Hippel Lindau (BGL)
BGL - moștenită ca o trăsătură autozomal dominantă, predispun-l la dezvoltarea tumorilor transportatorilor de multe țesuturi. Cu toate acestea, feocromocitom este caracteristic numai pentru pacienții cu diabet zaharat LGL 2. Spre deosebire de feocromocitoamele sporadice, în aceste cazuri, este mai puțin malign, rareori localizate în afara glandelor suprarenale, de cele mai multe ori apare pe ambele parti si la o varsta mai devreme. Vârsta medie a manifestărilor sale la pacienții cu tip LGL 2 - 28 ani, iar cele mai mici - 5 ani.
BGL are loc cu o frecvență de 2-3 cazuri la 100.000 de locuitori. Pacientii dezvolta adesea hemangioblastomul multicentricå (angiom) în retinei, cerebel si chisturile maduvei spinarii si a cancerului renal cu celule clare, multiple chisturi și tumori neuroendocrine non-funcționale ale pancreasului. Tumorile sac endolimfatic duce la pierderea auzului, amețeli și ataxie. Multe femei cu BGL in ovar, ligamentului larg al uterului, vaginului, colului uterin si labiile gasit chistadenomul probabil mezonefralnogo origine. La bărbați, ei sunt echivalentul chistadenomul a epididimului.
Pentru analiza genetică a primelor săptămâni de viață a copilului, sângele (într-o eprubetă cu EDTA) este trimis la un laborator unde ADN-ul limfocitelor pentru a investiga mutația genei VHL. Un copil născut într-o familie cu BGL, în căutarea de mutație deja cunoscut. Dacă bănuiți că un copil BGL dintr-o familie cu o mutație necunoscută se efectuează secvențierea directă a întregii regiuni de codificare a genei VHL și site-uri de îmbinare în căutarea de mutații punctiforme.
Diagnosticul clinic LGL este stabilit în acele cazuri în care pacientul dintr-o familie cu o mutatie cunoscuta in aceasta gena dezvolta una dintre tumorile lor, tipica a bolii. În lipsa unui istoric familial de BGL sale ar fi diagnosticată atunci când detectează oricare dintre două sau mai multe hemangioblastoamele, oricare dintre acestea, în asociere cu cancer de clar renale sau feocromocitom. BGL ar trebui să fie, de asemenea, suspectat la pacienții cu tumori multiple, manifestată într-o vârstă relativ tânără (mai puțin de 50 de ani pentru feocromocitom sau hemangioblastomul și mai puțin de 30 de ani pentru cancer renal cu celule clare). In astfel de cazuri, VHL secventierea genei.
Gene VHL (supresoare de tumori), localizată pe cromozomul 3 (Zr26-25 segment) codifică două proteine diferite constând din 213 și 160 resturi de aminoacizi. Ambele sunt implicate în factorii de distrugere induse de hipoxie (HIF-1 și HIF 2) - domenii ale acestor proteine se leagă la elongin și domenii - un HIF hidroxilat (reacție necesită prezența oxigenului). Acest complex se atașează apoi ubiquitin, care facilitează proteoliza intracelulare de HIF. Astfel, prezența oxigenului este distrus HIF. foame de oxigen a celulelor sau absența produselor funcționale ale genei VHL favorizează acumularea de HIE HIF sunt factori de transcripție care induc sinteza factorului de creștere vascular endotelial (EDF) al receptorului eritropoietinei eritropoietina, GLUT-1 și creștere derivat din trombocite factor-B. Toate aceste proteine sunt celule la hipoxie adaptate, dar abundența lor se crede, contribuie la dezvoltarea tumorilor.
Pacienții cu hemangioblastoamele de dezvoltare BGL, chisturi și cancer renal de clare, de obicei, necesită o mutație somatică rămase de tip sălbatic VHL gena alela (pierderea heterozygosity) sau hipermetilarea promotorului său (așa-numitul „al doilea hit“). Cu alte cuvinte, suficiente pentru dezvoltarea acestor tumori de acumulare HIF se produce numai atunci când ambele alele defecte genei VHL. Feocromocitomului BGL la pacienții cu tip 2 se dezvolta în starea normală a doua alele a genei. Prin urmare, sa sugerat ca unele mutatii missense in aceasta cauza dezvoltarea genei feocromocitomului, exercită influența mecanismul lor diferit, și anume - prin perturbarea legarea fibronectinei proteină sintetizată.
Ereditari mutații (germinative) genei VHL se găsesc în majoritatea familiilor cu BGL. Aproximativ 60% dintre aceste mutații sunt pierderea funcției (30% din proteinele trunchiate sunt sintetizate, iar 30% - au fost gena diviziune majoră), care provoaca BGL tip 1. Aproximativ 40% dintre pacienți sunt purtători de mutații missense care conduc la substituții de amino acizi din proteina sintetizat. În aceste cazuri, se dezvolta NGS diabet zaharat 2.
} {Modul direkt4
În 53% din cei 36 de pacienți cu LGL observate în Franța, prima detectare a tumorilor au fost doar feocromocitomului, care se manifestă adesea la o vârstă fragedă și în 42% din cazuri sunt bilaterale. În același timp, 11% dintre pacienți au fost identificate paraganglioma. feocromocitom maligne a avut loc în trei din 36 bolnyh- 18% din feocromocitomului a fost singura manifestare a bolii. Aproximativ 9% dintre pacienții cu feocromocitom unilateral (aparent sporadice) a demonstrat purtători mutațiile genei VHL germline. În unele regiuni ale Europei numărul pacienților este de 20%, ceea ce corespunde unui „efect fondator“ (înlocuind Tir98Gis în produsul genei VHL, așa-numita „mutatie Black Forest“ familii comune de origine germană).
Feocromocitom la BGL produc numai noradrenalinei și metabolitul său - Normetanephrine al căror nivel în plasmă, în astfel de cazuri, de obicei, crește. De aceea, la pacienții cu BGL de tip 2 (missense gena mutatie VHL) ar trebui să determine în mod regulat normetanephrine concentrația în plasmă.
Cel mai mare pericol pentru viața pacienților cu LGL este carcinom cu celule renale cu celule clare. Orice tumoare la rinichi solide, diagnosticat prin CT a abdomenului, pentru a fi eliminate. Chiar si chisturi in rinichi de astfel de pacienți sunt considerați a fi precanceroase, și eliminați-le mai bine. În cazul în care orice astfel de formațiuni la fiecare 6 luni, este necesar să se efectueze de înaltă rezoluție CT pentru a detecta semne de malignitate: o creștere a dimensiunii, chisturi pereți sau inegale partiții aspect în ele. mutații genetice Transportatorii Vhl necesită o monitorizare atentă.
- tip BGL 1. La tip BGL 1 feocromocitoamele nu se dezvoltă. mutațiile genei VHL ele conduc, de obicei, la pierderea funcției sale (ștergere deplasează cadrul de citire sau sinteza proteinelor trunchiate).
- BGL 2 diabet zaharat. În aceste cazuri, din cauza mutatii missense ale genei VHL, feocromocitom se dezvolta de obicei. diabet BGL 2 este împărțit în 3 subtipuri:
- tip BGL 2A (hemangioblastomul și feocromocitomul, dar risc redus de a dezvolta cancer renal de clar);
- Tip BGL 2B (hemangioblastom, feocromocitomul și un risc ridicat de a dezvolta cancer renal de clar);
- tip BGL 2C (fara pheochromocytoma hemangioblastoamele sau cancer renal).
tip Neirofibromatoz 1 (boala Recklinghausen)
Feocromocitomului caracteristic 0,1-5,7% pacienți Recklinghausen neurofibromatozå cu tipul 1 (NF-1). Cele mai multe dintre aceste tumori nu este detectat în timpul vieții, deoarece la autopsie la pacienții cu NF-1, frecvența lor de 3,3-13%. Ele sunt similare cu feocromocitom sporadice: în care sunt detectate 84% din cazuri tumora singur într-o singură glanda suprarenală, 10% - două fețe a tumorii la 6% dintre pacienți identificați paraganglioma extraadrenal și 12% - metastaza sau invazia țesuturilor înconjurătoare.
Feocromocitom gasit in 20-50% dintre pacienții cu NF-1 cu hipertensiune arterială. Prin urmare, în toate aceste cazuri, este necesar să se presupună prezența feocromocitom. cercetare corespunzătoare ar fi adecvată anuală generală toți pacienții NF-1, și în intervalele pentru a măsura tensiunea arterială și pentru a afla prezența simptomelor caracteristice feocromocitom (cefalee, transpirații, palpitații). Căutarea feocromocitom la pacienții cu NF-1 ar trebui să fie, de asemenea, în fața unei intervenții chirurgicale majore și sarcina.
Feocromocitom cu NF-1 se poate dezvolta la orice varsta - de la copilarie la starosti- varsta medie a pacienților la momentul diagnosticului este de 42 de ani. Aceste tumori ajung adesea dimensiuni enorme. Este surprinzător faptul că mulți pacienți, în ciuda secreția crescută de catecolamine și a tensiunii arteriale a rămas normală. Când SF-1 observată adesea coarctație a aortei și renal displaziei arterelor care pot provoca hipertensiune, feocromocitom simularea.
Recklinghausen bază mutația bolii constă NF-1 (supresoare de tumori), localizată pe cromozomul 17 (segmentul 17q11.2) și neyrofibromin care codifică (proteină din 2818 de resturi de aminoacizi), care inhibă proteinele Ras. Neyrofibromina absenta duce la dezvoltarea tumorii. NF-1 este o tulburare autozomal dominanta (desi 50% din cazuri par a fi sporadice) - frecvența este de aproximativ 300-400 de la 1.000 de locuitori 000. Diagnosticul este de obicei în copilărie sau adolescență, pe baza clinica, cu toate că este posibil și analiza genetică.
Copilul SF-1 se manifestă, de obicei, glioame ale nervului optic, ceea ce duce la tulburări vizuale, și ca un adolescent - neurofibroamele plexiform. Pacienții apar neurofibromas subcutanate și Schwannoame rădăcini ale nervilor cranieni si spinali. De multe ori există anomalii ale scheletului. hamartoame hipotalamice poate provoca dezvoltarea sexuala prematura. Uneori există iris hamartoame (noduli Lisch). risc crescut de a dezvolta alte tipuri de tumori (în special tumori maligne ale membranelor nervilor periferici) și leucemii (leucemie granulocitară special juvenilă cronică). pistrui Caracterizat subrat si falduri ale pielii. Există mai multe pete de culoare de cafea cu lapte, numărul și mărimea care cresc odată cu vârsta. Majoritatea pacienților care au mai mult de șase astfel de spoturi cu margini netede și 1,5 cm în diametru.
mutatii NF-1 purtători de gene necesită o monitorizare atentă.
Familie sindroamele / feocromocitoamele paraganglia: complex mutațiile genei suktsinatdegidrogenaznogo
Membrii unor familii dezvolta adesea Paragangliomul multicentrice ale capului și gâtului, paraganglioma simpatic și feocromocitom suprarenale. Baza acestor sindroame sunt trei dintre cele patru mutații ale genelor care codifică complexul mitocondriale II, care cuprinde SDGV și SDG SDGD. Într-o familie olandeză cu sindromul descoperit o mutatie a unei gene situate pe cromozomul 11.
La toti pacientii cu mutatii SDGV genelor SDG si SDGD (alele paterni) predispoziție de a dezvolta paraganglia moștenite în mod autosomal dominant. Atunci cand genele mutatii evolua SDGV SDGD si, de asemenea, feocromocitomul suprarenale. La persoanele cu mutatii genetice apar Paragangliomul SDGD numai în cazurile în care gena mutantă este moștenită de la tatăl său. Purtători genei LDH mutațiilor germinale este de aproximativ 12% dintre pacienții cu aceste sindroame.
paraganglia Frecvența capului / gâtului este 10-33 cazuri la 1 milion de locuitori. Într-o serie de observații care au inclus 34 de pacienti cu astfel de tumori, mutatii genetice germene LDH au fost gasite in 41% din cazuri. Dintre pacientii cu aceste mutatii in 79% din cazuri au avut mutatii genetice SDGD, iar restul - mutatii genetice SDGV.
După cum sa menționat deja, toate aceste sindroame sunt cauzate de mutații ale genelor nucleare care codifică trei dintre cele patru subunități care alcatuiesc complexul mitocondrial II (SDH), care în ciclul Krebs oxidează succinat la fumarat. Oligomeric LDH include SDGA (având flavoproteina o greutate moleculară de 70 kDa) SDGV (proteină fier-sulf, 30 kDa), SDG (subunitate a citocromului b, 15 kDa) și SDGD (o altă subunitate a citocromului b, 12 kDa). Componentele genei LDH cuprinzând b (SDG și SDGD) localizate în membrana mitocondrială și este atașat subunitate SDGA SDGV care transportă lanț de electroni în transferul de electroni Coenzima Q (ubiquinone). SDG este necesară pentru ciclul și energie producția de acid citric în condiții aerobe. Defecte genetice SDG viola funcția mitocondrială, cauzand celulele hipoxice. Aceasta crește secreția factorului de creștere endotelial vascular (FED) necesare pentru progresia tumorii. (Mutatii embrionare SDGA provoca dezvoltarea corpului cromafin nu este, și dacă boala - boala fatala copilariei neurodegenerative cauzate de patologii mitocondriale).
- gena Mutațiile SDGD. gena SDGD localizată pe cromozomul 11 (q23 segment). Mutațiile ale acestei gene au fost găsite în majoritatea pacienților cu paraganglioma familială. În familiile cu predispoziție la astfel de mutații de dezvoltare feocromocitoamele paraganglia și înregistrate numai în caz de moștenire a genei mutante de la tată ( „imprimare genomica materne“). copii de sex masculin care au primit gena mutantă de la mama SDGD, nu se imbolnavesc, dar pot trece gena (și, prin urmare, o predispoziție la dezvoltarea corpului cromafin) urmașilor lor. Femeile care au primit gena mutantă de la tata, Paragangliomul dezvolta, dar copiii lor - nr. La persoanele cu mutatii germline in SDGD gena special adesea dezvolta tumori Ganglionii parasimpatici (embrionic asociat cu corpul cromafin simpatic), în zona capului și gâtului (angioneuroma). Din moment ce acestea paraganglioma aproape nu secretă catecolamine, ele sunt numite „non-cromafin“. Angioneuroma dezvolta, de obicei, dintr-un glomus somnoros la bifurcatia arterei carotide (chemodectoma) sau la baza craniului și sunt unice sau multiple, simple și duble. Toți acești pacienți au nevoie de studiu atent al zonei gâtului, scanarea și monitorizarea regulată pe tot parcursul vieții. Aproximativ 74% dintre pacienții cu paraganglioma cauzate de mutatii gena SDGD detectate tumori multicentrice, iar 50% dintre pacienții cu feocromocitom și germline mutatii aparent izolate gena sunt ascunse SDGD paraganglioma. Astfel, în toate aceste cazuri, este necesar să se caute alte paraganglioma folosind RMN si / sau tomografie cu emisie de pozitroni (PET). În familiile de origine olandeză detectate două „fondator mutatie“ gena SDGD: Ley95Pro și Asp92Tir. „Mutația fondatorului“ (Gln109H), de asemenea, găsite în familiile de origine italiană. Probabilitatea de a dezvolta corpul cromafin (penetranta) în purtători ai genei mutante SDGD crește vârsta paterni de la 33% la vârsta de 30 până la 83% în 60 de ani. Paraganglioma, cauzate de mutatii ale genei SDGO sunt rareori canceroase. Cu toate acestea, paraganglioma de col uterin poate metastaza la limfatici regionali noduri, plămânii și kosti-, uneori, timp de mai mulți ani nu se manifesta clinic. Poate că dezvoltarea corpului cromafin și în alte locuri, precum și feocromocitoamele care necesită o monitorizare continuă a acestor pacienți.
- gena Mutațiile SDGV. gena SDGV este mapat la cromozomul 1 (1r36 segment). Mutatii ale acestei gene sunt legate cu paraganglioma mai mică decât gena mutatie SDGD. Tumorile în aceste cazuri, se poate dezvolta oriunde pe sistemele parasimpatic si simpatic - de la gât la pelvis, precum si in glandele suprarenale (feocromocitom), dar Glomus somnoros în gât vin mai puțin frecvent decât cu mutații a genei SDGD. La mutații genetice embrionare SDGV metastaze paraganglia la momentul diagnosticului sunt detectate mai des (35% din cazuri) decât în mutațiile genei SDGD germinale. Calculat penetranta SDGV genei mutante este de aproximativ 31%. Astfel de pacienți sunt mai susceptibile de a dezvolta alte tipuri de cancer. Dintre cele 53 de pacienti cu mutatii in aceasta gena au fost detectate două carcinom cu celule renale cu celule clare și unul - cancer tiroidian papilar.
- gena Mutațiile SDG. gena SDG este localizată pe cromozomul 1 (q2 segment). Cand mutatii ale acestei paraganglioma gena detectată într-o singură familie europeană, iar toți pacienții au fost localizate în afara glandelor suprarenale. Astfel, toți pacienții cu paraganglioma și feocromocitom trebuie să își asume prezența mutațiilor germinale ale genelor care codifică complexul SDH. Analiza genetică este indicat în special în cazurile paraganglia gâtului (în care mutațiile germinale sunt mai mult de 15% dintre pacienți), sau feocromocitomului paraganglia multifocale, corp cromafin sau antecedente familiale de feocromocitomului și pacienți paraganglia de origine olandeză.
Alte sindroame genetice, inclusiv feocromocitom
- Carney triada. Paragangliomul multicentricå apar la pacienții cu triada Carney (a nu se confunda cu complexul Carney!). Acest sindrom apare de obicei la femeile mai tinere de 40 de ani și include paraganglioma ascunse de stomac leyomiosar-comă și condrom pulmonare.
- Sindromul Beckwith-Wiedemann. Feocromocitom (inclusiv ambele părți) au fost observate la pacienții cu BWS. Pacienții au fost observate și alte încălcări, în special - hipoglicemia nou-nascuti, hernie ombilicală, hernie cordonului ombilical, macroglosia si gigantism, precum și un risc crescut de tumori maligne.
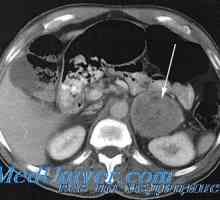 Feocromocitom: clinică, mecanisme patologice de dezvoltare
Feocromocitom: clinică, mecanisme patologice de dezvoltare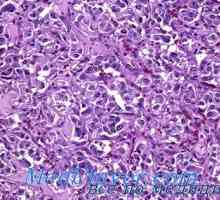 Diagnosticul, diferențierea și tratamentul feocromocitomului
Diagnosticul, diferențierea și tratamentul feocromocitomului Hiperfuncția medulosuprarenalei (feocromocitomul) morfologia și anatomia patologică
Hiperfuncția medulosuprarenalei (feocromocitomul) morfologia și anatomia patologică Simpatogoniomy și feohromoblastomy. corpurile se schimbă cu feocromocitom
Simpatogoniomy și feohromoblastomy. corpurile se schimbă cu feocromocitom Efectul de mutații în Ret eficacitatea tratamentului cancerului tiroidian medular
Efectul de mutații în Ret eficacitatea tratamentului cancerului tiroidian medular- Eterogenitate intratumorală determină rezultatul tratamentului cancerului
 Cauzele și patogeneza feocromocitom
Cauzele și patogeneza feocromocitom Simptomele feocromocitom la copii
Simptomele feocromocitom la copii Simptome și semne de feocromocitom
Simptome și semne de feocromocitom Diagnosticare si analiza cu feocromocitom
Diagnosticare si analiza cu feocromocitom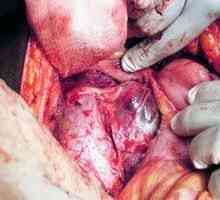 Suprarenală feocromocitom - o tumoare malignă, boala, clinica fotografie, cod în conformitate cu…
Suprarenală feocromocitom - o tumoare malignă, boala, clinica fotografie, cod în conformitate cu…- Simptome feocromocitom, diagnosticul și tratamentul feocromocitomului
- Hipertensiune suprarenale
- Sindromul de neoplazice endocrine multiple tip 1
- Feocromocitom, o boală cauzată de o tumoră benignă sau malignă a țesutului cromafin adrenal sau…
- Distinge> tumori suprarenale benigne si zlokachestvennyeref = „des204.htm“,…
- Sănătate Enciclopedia, boli, medicamente, medic, farmacie, infecție, rezumate, sex, ginecologie,…
 Feocromocitomul și paraganglioma: simptome, semne, tratament, cauze
Feocromocitomul și paraganglioma: simptome, semne, tratament, cauze Cancer tiroidian medular
Cancer tiroidian medular Sindromul neoplaziei endocrine multiple, de tip ii (Maine 2): cauze, simptome, tratament, simptome
Sindromul neoplaziei endocrine multiple, de tip ii (Maine 2): cauze, simptome, tratament, simptome Stările asociate cu supraproducție de hormoni suprarenali
Stările asociate cu supraproducție de hormoni suprarenali
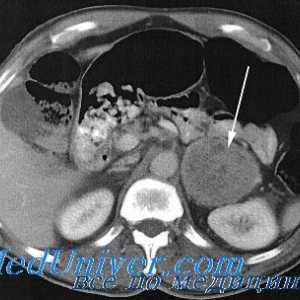 Feocromocitom: clinică, mecanisme patologice de dezvoltare
Feocromocitom: clinică, mecanisme patologice de dezvoltare Sindromul neoplaziei endocrine multiple, de tip ii (Maine 2): cauze, simptome, tratament, simptome
Sindromul neoplaziei endocrine multiple, de tip ii (Maine 2): cauze, simptome, tratament, simptome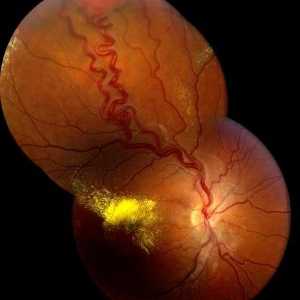 Von Hippel-Lindau bolii: simptome, prognostic, tratament, cauze, simptome
Von Hippel-Lindau bolii: simptome, prognostic, tratament, cauze, simptome Simptomele feocromocitom la copii
Simptomele feocromocitom la copii Diagnosticare si analiza cu feocromocitom
Diagnosticare si analiza cu feocromocitom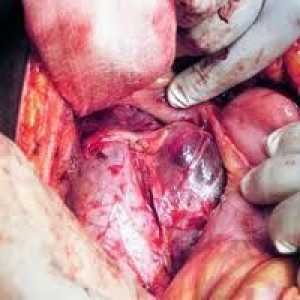 Suprarenală feocromocitom - o tumoare malignă, boala, clinica fotografie, cod în conformitate cu…
Suprarenală feocromocitom - o tumoare malignă, boala, clinica fotografie, cod în conformitate cu…