Trenaunay Sindromul Klippel-Weber. Sindromul Larsen la făt
atunci când acest lucru sindromul
conținut
Sinonime. Sindromul Klippel-Trenaunay (Klippel-Trenaunay) și sindromul angioosteo-hipertrofie.
Prevalența. Rare.
Conform ipotezei R. Happle tip paradominantny de moștenire poate explica natura bolii cel mai satisfăcător. Heterozigot pentru persoane fizice defect monogenice sunt fenotipic normale. Boala poate avea loc numai în cazul în stadiile incipiente ale embriogenezei apare mai mutației somatice a alelei normale a genei. In astfel de cazuri, embrionul va avea mosaicism cu prezența liniilor celulare homozigote sau mutații heterozigote în ceea ce privește datele. Acest lucru explică distribuția localizată a defectelor detectabile clinic.
riscul de recurență. Poate să lipsească, cu toate acestea, având în vedere un mod autozomal dominant de moștenire, în unele cazuri, riscul poate crește.
diagnosticare. Ultrasonografia pot fi detectate hydrops fetale (din cauza insuficienței cardiace), edem al membrelor și hipertrofia țesuturilor moi (în plus față de lungimea circumferențiară a indicatorilor de mărime), ascită, hemangiomatoza patologice volum LARG localizarea formării intraperitoneal și hepatomegalie. Un caz de un diagnostic corect cu ajutorul ultrasunetelor tri-dimensionale.
patogenia. Anomalii ale capilare formele atipici ale venelor varicoase și malformații vasculare venoase, care conduc la formarea unor formațiuni în vrac limitate.
tulburări genetice. Probabil, cauza este un defect monogenica este localizat pe brațul lung al cromozomului 5q sau pe 5r11 său braț scurt.
anomalii asociate. Boala poate fi complicată de dezvoltare-Merritt sindromul Kasabaha (Kasabach-Merritt), care este însoțită de trombocitopenie, cauzată de trombocite hemangioame țesuturi de consum, precum și simptome de insuficiență cardiacă.
diagnostic diferențial. Limfangioma și sindromul Proteus (Proteus).
perspectivă. Atunci când detectat în perioada prenatală, boala este de obicei mai severă cu prognostic nefavorabil, mai ales dacă este combinat cu dezvoltarea insuficienței cardiace.
tactici obstetrică. In formele severe ale sindromului poate fi cerut la avort, în alte cazuri, tactici obstetricale de sarcină nu se schimbă.
Sindromul Larsen la făt
atunci când acest lucru sindromul marcat statura scurt, luxație congenitală multiplă a articulațiilor și caracteristicile faciale anormale. Există două forme ale sindromului: letale și non-letale. Mortalitatea în prima formă a bolii provocate de hipoplazie pulmonară.
răspândire. Rare.
Video: SOS! O situație teribilă! Sashenkin picior imens și este în pericol!
etiologie. Formarea de colagen anormale, care se caracterizează prin reducerea exponent împotriva E.-1 / 01-2 lanțuri de colagen tip I.
diagnosticare. Anomaliile articulații dislocații (luxație de șold, genunchi unghi de flexie deschis anteriorly datorită prezenței oaselor suplimentare clubhand carpian, etc.), picior strâmb poate fi combinat cu scurtarea Rizomelicheskaya superioare fibula hipoplazie membrelor și uneori cu corpuri vertebrale despicătură frontale. Anomaliile feței în acest sindrom includ nas profund, frunte proeminente și hypertelorism.
tulburări genetice. Acesta a raportat ca autozomal recesiva tipuri dominante sau autosomal de moștenire. Gena în care mutația este localizată pe cromozomul -r14.1 Zr21.1.
diagnostic diferențial. În lipsa unui istoric familial de astfel de pacienți stabilesc de obicei artrogipoza diagnostic.
Video: Dasha Foltz 10 luni
perspectivă. detectarea în timp util și susținerea măsurilor pentru corectare traheomalacia, insuficienta pulmonara si instabilitate vertebrala (sub forma de cifoza din cauza hipoplazia severe de una sau două corpuri ale vertebrelor cervicale, de obicei, IV sau V), pot îmbunătăți prognosticul, deși acest lucru este posibil doar cu o formă non-letale .
 Cauzele anomaliilor fetale. Riscul de apariție a malformațiilor fetale
Cauzele anomaliilor fetale. Riscul de apariție a malformațiilor fetale Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus
Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus Amputarea congenitală a fătului. Sindromul aglossia-adactylia
Amputarea congenitală a fătului. Sindromul aglossia-adactylia Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale
Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale Akromezomelicheskaya displazie. sindromul eykardi
Akromezomelicheskaya displazie. sindromul eykardi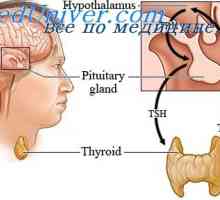 Hipofosfatazia. hipoplastic stang
Hipofosfatazia. hipoplastic stang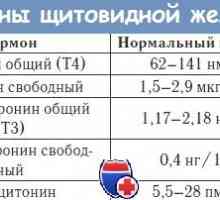 Sindromul Holt Oram. sindromul Gidroletalny
Sindromul Holt Oram. sindromul Gidroletalny Mikrotsefalichesky nanism primar. Sindromul dezordonate-Lax
Mikrotsefalichesky nanism primar. Sindromul dezordonate-Lax Sindromul Noonan într-un fat. oligohidramnios Sequence
Sindromul Noonan într-un fat. oligohidramnios Sequence Osteogeneză imperfectă. Diagnosticul și prognosticul osteogenezei imperfecta la făt
Osteogeneză imperfectă. Diagnosticul și prognosticul osteogenezei imperfecta la făt Sindromul Pfeiffer. Diagnosticul și prognosticul sindromului Pfeiffer
Sindromul Pfeiffer. Diagnosticul și prognosticul sindromului Pfeiffer Alele dominante și recesive de cromozomi. dominantele autozomal
Alele dominante și recesive de cromozomi. dominantele autozomal Anomalii ale organelor genitale feminine. Sindroame Kaufman-mac-Cusick și…
Anomalii ale organelor genitale feminine. Sindroame Kaufman-mac-Cusick și…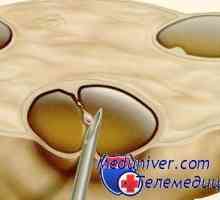 Diagnosticul genetic preimplantare (PGD). Indicații și posibilități
Diagnosticul genetic preimplantare (PGD). Indicații și posibilități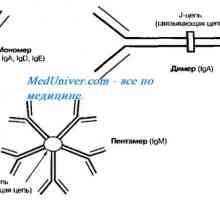 Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation
Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation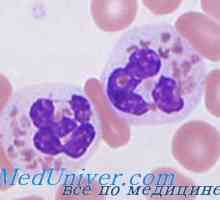 Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann
Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann Sindromul sturzh Weber
Sindromul sturzh Weber- Sindromul de neoplazice endocrine multiple tip 1
- Teleangiektaticheskaya Ataxia (sindromul Louis-Bar), este o boală relativ rară, cu modul autosomal…
 Sindromul rezistenței la hormon tiroidian
Sindromul rezistenței la hormon tiroidian Sindroame monogenice mvpr
Sindroame monogenice mvpr
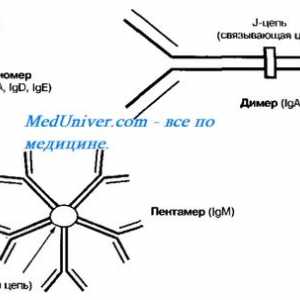 Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation
Autozomal sindromul hyperproduction recesiv de imunoglobuline m (lgM). gena ajutor de Mutation Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann
Neutropenie ereditara. Sindromul neutropenie congenitală severă Kostmann Sindroame monogenice mvpr
Sindroame monogenice mvpr Diagnosticul genetic preimplantare (PGD). Indicații și posibilități
Diagnosticul genetic preimplantare (PGD). Indicații și posibilități Akromezomelicheskaya displazie. sindromul eykardi
Akromezomelicheskaya displazie. sindromul eykardi Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus
Sindromul de gudron la făt. Sindromul Aase, Holt-Oram fetus Osteogeneză imperfectă. Diagnosticul și prognosticul osteogenezei imperfecta la făt
Osteogeneză imperfectă. Diagnosticul și prognosticul osteogenezei imperfecta la făt Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale
Vater de asociere la făt. Sindromul polidactilie și goldenhara fetale Alele dominante și recesive de cromozomi. dominantele autozomal
Alele dominante și recesive de cromozomi. dominantele autozomal